- A+
A:请问分子对接后还可以调整侧链方向吗?
殷赋科技:调整氨基酸侧链?可以的啊,用你熟悉的软件,比如MOE,调整后对该氨基酸残基和附近若干受影响的残基(以及配体,如果配体有参与的话)进行能量优化,其余部分保持固定。
A:这个是模拟后的结果,但是我觉得天冬氨酸残基应该偏向钙离子。

下面是手动调整之后的侧链摆向,这样之后还需要继续模拟是吗?
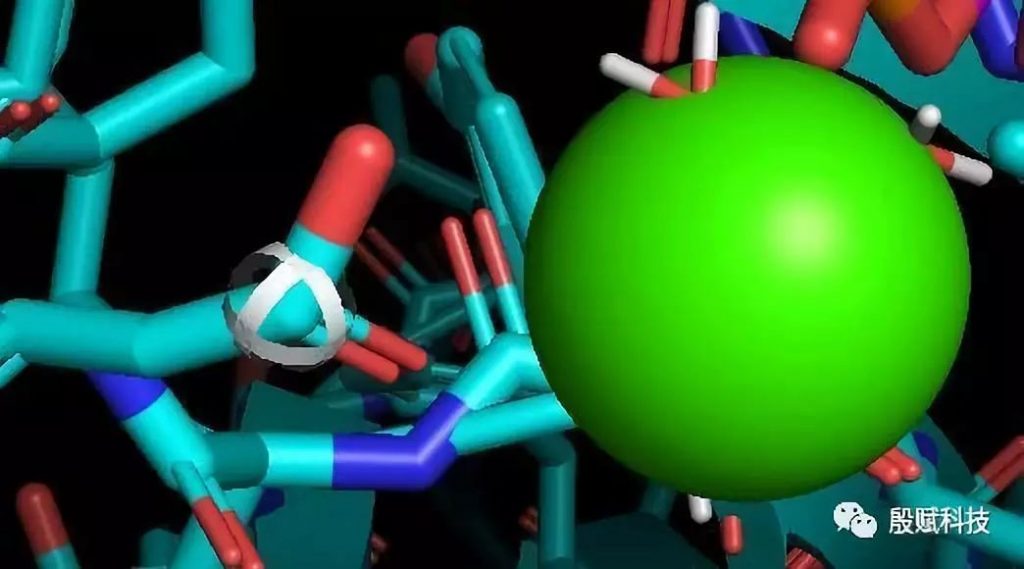
殷赋科技:这个天冬氨酸应该是跟钙离子形成配位键的吧,在对接之前就应该把这部分弄好。调整后怎么做,我上面说了方法。
A:分子对接大分子和蛋白,两者之间以何种化学键结合更好?激动剂和抑制剂的要求不一样吧?
殷赋科技:两者以何种化学键结合更好,这个问题无解。要先搞清楚激动剂和抑制剂的机理,有时候它们可能是同一个位点、是竞争关系,有时候是不同位点(别构位点)。这个不能在计算上给出答案,只能通过实验研究去了解机理,但是,计算可以协助解释(比如做分子动力学模拟,研究激动剂和抑制剂/拮抗剂的结合模式区别)。所以,基本上,脱离了实际情况,这个问题无解。
A:哦谢谢张老师,我之前做了小分子与一个疾病的靶点的对接,大多是以范德华力和氢键结合,是与蛋白残基结合越多越好还是?不太懂,张老师能都推荐一些资料。
殷赋科技:结合越多,打分通常是越好的,但未必实际的生物活性越好,因为打分跟生物活性的相关性不大。一般的作用力都是疏水作用和氢键。如果足够好(打分好、结合模式好),就可以考虑做实验验证了。参考资料:《如何合理对待分子对接结果》。
B:请问从大分子和小分子对接出来的结果,怎么样才能知道相互之间是什么作用力呢?比如怎么知道哪个是氢键或者疏水或者静电作用力。
殷赋科技:你用什么软件做的?商业软件一般都有工具可以显示相互作用,我们平台可以做对接,做完就有相互作用分析,作用力类型是最全面的。
B:Autodock。
殷赋科技:那就用DS来显示吧。我推荐用我们平台的Dock6方案,计算准确,分析功能齐全,作用力图可以直接下载下来发文章,又或者按照这篇文章用pymol来做图。
C:张老师,如何能够快速有效的保留关键氨基酸残基?因为受体太大了,我想把不太关紧的去处,但是方法不得当,经常删到有效的残基。
殷赋科技:经常删到有效残基,难道你要操作很多不同的蛋白么?不是的话,那我怀疑是操作问题。用DS、MOE之类的软件,将要保护的残基选上,然后反选。或者用我们平台,在定义受体的时候可以选择你要保留的残基作为受体那部分,或者干脆就给程序自己去处理,它会以口袋中心为中心,删掉30A以外的残基,这样保证蛋白不太大,但避免“伤及无辜”。而对接后用来分析的蛋白,依然是完整的蛋白。
C:谢谢,我再试一下平台。
D:有哪位老师知道如何在pymol中给配体显示电子云密度?
E:你是说xray的电子密度图吗?还是分子轨道理论里的电子出现的概率: 概率密度?
D:不是,自己设计的小分子对接到蛋白里,就a图里的样子。
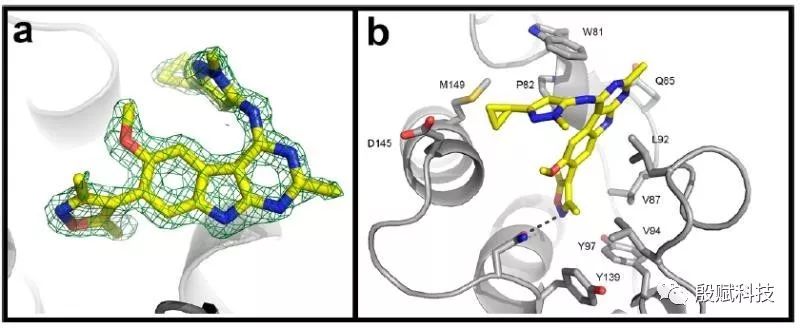
E:这个是单晶xray实验数据,在rcsb上有下载链接,这个不是计算出来的。
F:@D选中配体,show mesh。
A:薛定谔分子对接软件。如何批量修改导入分子的名字?
B:用EditPlus(共享)或者CrimsonEditor(开源/免费)这样的文本编辑软件。
Edit菜单下,有类似Find in files之类的功能,也就是对某目录下的多个文件的内容进行“查找、修改”。
一般是用于编程时,改变某个变量的名称,这个名称往往分布在多个源代码文件中。
如果是简单的“替换”,那还比较容易。
如果类似您这种情况,那还要用到“正则表达式”
------------
有个取巧的办法:
即把
# Name: 1
替换为
# Name: m1
-----------
话说回来,用文本编辑器,修改200个文件的内容,手工操作,10分钟而已。
殷赋科技:@A这个问题还是笼统,举例说明从怎样修改成怎样才能帮到你。
A:@殷赋科技 sdf导入的名字都一样。后面查找的时候有点乱。希望能够在这里或是导入前改变下名字。

殷赋科技:这个名字是分子的内部名,修改下就可以了,那你想改成什么?简单的编号1、2、3?还是其他名字?只要是有规律的,用Linux命令、python脚本都可以办到。
导入前是什么格式的文件?sdf和mol2文件格式不同,分子名的位置不同。
A: sdf,在sdf文件里面标柱或是不标注都没有用,用chembiodraw画出来的。
殷赋科技:你是画出来保存为sdf,然后再导入薛定谔吧,你可以改一下几个名字,看看有没有影响。是不是这些名字是文件名?
A:对的。我在结构下面标注名字没有用。
殷赋科技:那你可以将各分子分裂出来,成为一个一个文件,每个文件给予不同的编号。
A:我试试。主要是自己设计出的分子上千。
殷赋科技:用Linux命令。
A:目前还没有用过命令。
殷赋科技:假设文件名是test.sdf,里面包含1000个一下的分子(即两位数)。
分割成每个文件的格式为split-*.sdf,*是数字编号,比如split-0.sdf, split-1.sdf。
那么,Linux命令是:csplit test.sdf /\\$/+1 {*} -n2 -f split -b "-%d.sdf"。
产生的结果是,第一个分子是split-0.sdf,第二个是split-1.sdf,以此类推,最后一个文件是空文件,可以删掉。
注意命令中的空格和符号,不熟悉命令的人容易出错。
4
A:有一条多肽,被拆分成两段,然后在体内通过实验方式让拆分的两段靠近,并正常行使功能。有做过类似实验的吗?原文是利用tn7转坐插入的方式,筛选了几个候选位点,我的问题是有没有一种通过预测的方式提前确定多肽有哪些可以拆分的点?
殷赋科技:如果知道它的靶标(蛋白),可以做对接、动力学,找到有关键作用的“点”。
A:意思是切割多肽与配体结合的活性位点?
殷赋科技:将多肽与靶标蛋白对接,看看它是怎么结合的,确定多肽上关键的点。
A:一直不太会看受体配体结合的图怎么分析,怎么确定关键点,凭经验吗,有没有相关教程可以推荐一下呢。
殷赋科技:用我们平台就可以了,对接完就有相互作用分析,会显示出有哪些作用力。
A:有受体配体结合的图,不知道怎么确定关键点,刚接触不久。
殷赋科技:也可以用DS来显示,但没有我们平台显示得全面。有相互作用的地方就是关键的点咯。你都有结合图了,用DS显示一下不就好了,主要看氢键吧。这个它能显示出来。
A:多问一句,pymol能不能达到同样的展示效果。
殷赋科技:pymol可以显示氢键。
B:@A 蛋白质的环重排。
A:一般像抗有可变区,发生基因重排,能简单解释一下蛋白质的环重排是什么意思?@B跟找蛋白质序列拆分位点有什么关系,或者有类似文献可以推荐一下。
B:我是说你的改造思路有点像环重排,你可以搜一下有人将荧光蛋白拆分后,两个片段重新在体内靠近时还会发荧光。
A:嗯,是的,这个了解,现在是在想通过预测的方式找多肽的拆分位点,然后去验证,两个片段空间上靠近也能行使功能。
B:有结构的话你试试在loop区域切割。我没见过类似的预测方法,要么依靠直觉,要么依靠随机重排。
A:荧光蛋白确实拆分的比较多,有许多这样的专利。
B:如果是简单的环化是有一些算法与数据库,具体你要查一下。
A:好的,我再去查一查,谢谢。



