- A+
现代药物的发现是一个数据驱动的过程,其中每一个阶段几乎都需要大量数据来辅助决策。药物设计者借助科学思维,使用信息技术对数据进行处理,以揭示药物、靶标和疾病相互之间的关联,形成系统化的药物信息。因为这些数据涉及到类似靶标序列、三维结构、生物活性和药代动力学性质等信息,所以药物信息可以说是化学信息和生物信息的总和。一般而言,化学信息处理的对象是小分子,而生物学信息处理的对象为基因和蛋白质等大分子。两者相互依存、相互影响。
01
化学信息处理
化学信息学是一门新兴的、多学科交叉的边缘学科,它运用数学、统计学与计算机程序设计的方法,对化学信息进行分析和管理。下面我们对化学分子结构表达、分子结构的数学描述、分子相似性分析、预测模型构建以及虚拟化合物库设计展开介绍。
化学信息学在药物设计中的应用
1)化学分子结构表达
化学分子由原子和化学键组合而成,它们之间通过化学反应进行转换。因此化学信息包括文本、数据、图形结构及其性质和反应的相关信息。通常,我们从多个层面对化学小分子进行表达,其中包括一维线性表达、二维和三维结构式表达。一维线性结构在面对大量化合物结构进行快速储存、读取和操作时,具有较大优势。常见的线性符号表示法有SMILES、SLN和ROSDAL等几种。二维矩阵表达主要针对ChemDraw、ChemSketch、JME Editor等分子编辑软件生成的以图形模式存在的分子结构式,利用矩阵运算对化合物结构进行灵活处理。
2)分子结构的数学描述
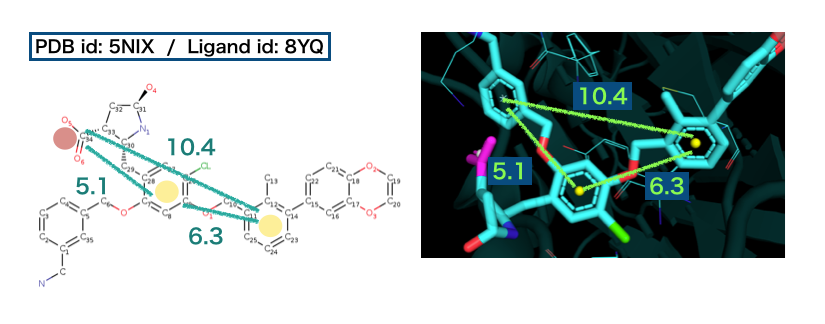
分子结构可以借助分子结构表达式存储到计算机上,而分子信息处理则需要通过分子结构的数学描述去实现。分子描述符是指分子在某一方面性质的度量,既可以是分子的物理化学性质,也可以是根据分子结构通过各种算法推导出来的数值指标。

3)分子相似性计算
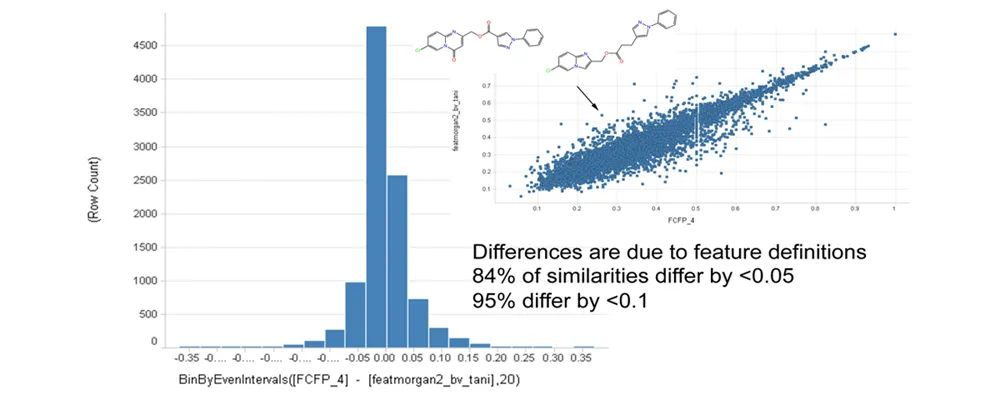
在化学信息中,分子相似性是非常重要的概念。“结构相似的分子一般具有相似的理化性质或生物活性”是药物化学中的共识。一般来说,先导化合物的发现阶段,更注重分子的多样性;而先导化合物的优化阶段,则更多考虑化合物的相似性。相似性分析可以简化为三步:首先,将分子用适当的方法进行描述;其次,选择合适的相似性测量方法,以便定量测定一堆分子之间的相似性;最后,采用算法进行相似性计算。根据分子相似性原理,我们可以在数据库中搜索与目标分子具有一定相似性的分子,由此演变的相似性搜索是现在许多虚拟筛选技术的理论基础。
4)虚拟化合物库设计
化合物库通常是指用于高通量筛选的化合物集合,包括真实的和虚拟的化合物库。研究人员按照一定规则去设计虚拟化合物库,然后进行虚拟筛选,之后再合成少量具有成药前景的化合物样品,并进行生物实验筛选,以提高活性化合物的发现比例。按照用途的不同,化合物库通常可以分为用于先导化合物发现的多样性库(diverse library)和用于先导化合物优化的靶向库(targeted library)。设计虚拟靶向库的步骤包括:片段库构建、核心骨架确定、组合靶向库构建、结构优化和评价。
02
生物信息处理
在药物设计中,我们借助生物信息学的知识系统,利用基因组中编码区的信息进行蛋白质的结构预测和功能预测,并将此类信息与生物体和生命过程的生理生化信息相结合,阐明其分子机理,进而对蛋白质和核酸进行分子设计、药物设计。
从这里可以获得生物大分子序列、结构、功能等相关数据:
1)UniProt(http://www.uniprot.org/);
2)Protein Data Bank(http://www.rcsb.org/)(PDB库中存在同一蛋白具有不同分辨率的结构,或者同一蛋白质与不同配体结合形成复合物结构等情况,需注意);
3)KEGG(http://www.kegg.jp/kegg/)。
以上数据库详见第五篇文章《跟我学药物设计 | 药物和靶标的相互作用》。
生物信息学在药物设计中的应用
序列分析
序列分析包括从基因组序列中获取信息的核酸序列分析和蛋白质序列分析。药物设计中,最常见的是蛋白质序列分析。它主要有两类:单个蛋白质序列分析和多个蛋白质序列分析。其中,后者常用于研究由共同祖先进化而来的序列。
蛋白质结构预测
研究蛋白质的目的在于了解蛋白质的作用,它是如何行使生物学功能的,以及它与其他分子之间的相互作用。虽然蛋白质由氨基酸的线性序列组成,但是它只有折叠成特定的空间构象才能具有相应的活性和生物学功能。通过对蛋白质的结构分析,确认功能单位或者结构域,可以为遗传相关的基因工程技术提供目标,为设计新的蛋白质或改造已有蛋白质提供可靠的依据,同时为药物分子提供合理的靶点结构。迄今已知的蛋白质序列数目庞大,但实测结构少得可怜。为了解决结构数量与序列数量之间的巨大差距,我们通过理论计算方法,从氨基酸序列出发,进行蛋白质一级、二级和三级结构的预测。相关知识见《跟我学药物设计 | 靶标的主要类型与结构特征》。
同源模建
同源建模在药物的发现过程中被广泛用来预测靶标蛋白质的三维结构。基本流程包括:数据库搜索与模板选择、二重或多重序列比对、保守区主链骨架构建、模型优化以及结构合理性评估。蛋白质序列比对和结构预测在药物研发过程中具有十分广泛的应用,它可以帮助研究者进行基于结构的药物设计,或者采用分子动力学模拟方法了解蛋白质的动态结构特征。
从头预测
从头预测法主要采用理论计算(如分子力学、分子动力学计算)方法,根据物理化学的基本原理,不依赖已知结构的同源相似物信息,直接从理论上预测一个序列对应的蛋白质三维空间结构。对于没有同源模板或者同源性过低的序列,无法采用同源模建方法进行蛋白质结构预测。此时,可采用从头预测的方法,其主要思想是基于能量计算,寻找能量最低点时的折叠模式。这种方法计算量巨大,一般可以预测的蛋白氨基酸数目较小。近些年来,利用人工智能、数据科学和生物信息学,蛋白质结构预测领域有了突破性进展。2021年7月开源的AlphaFold2对于大部分蛋白质结构具有高度准确的预测精度,可媲美实验方法【1】。
常用的蛋白质结构预测工具
1)I-TASSER

一款可以在线使用的蛋白预测工具。其原理是先进行多模板搜索,然后基于多模板分别建模,最后进行结果整合。没有模板的部分则采用从头预测的方法来填补。I-TASSER可以预测最多1500个氨基酸的蛋白质分子,精度很高。
2)AlphaFold
Google旗下DeepMind开发的一款人工智能程序,它使用深度学习算法通过蛋白质序列来预测蛋白质结构。近期开源的Alphafold2对大部分蛋白结构的预测精度就达到了空前的准确度,“不仅与实验方法不相上下,还远超解析新蛋白质结构的其他方法”,多数预测模型与实验测得的蛋白质结构模型高度一致。
3)RoseTTAFold
一款人工智能软件系统,受AlphaFold2设计思路的启发,RoseTTAFold利用神经网络技术综合一维、二维、三维以及交互信息,推断出更为准确合理的折叠方式,同时还能预测蛋白质复合体的结构。RoseTTAFold对蛋白质结构的预测精度已与AlphaFold2不相上下,而它对硬件设备的要求较低,仅需一块RTX2080显卡,便可以在10分钟内计算得到400个氨基酸残基以内的蛋白质结构。
03
信息计算新技术
随着互联网时代信息与数据的爆发式增长,科学、工程和商业计算领域海量数据的处理对计算能力的需求激增。为了解决这些问题,集群计算、网格计算、云计算涌现于世。
常用的信息计算技术
集群计算
集群计算关注的是计算资源,它将一组相互独立的、通过高速网络互联的计算机软件或硬件连接起来高度紧密地协作完成计算工作,即将互联网中的PC机或工作站连接起来,提供廉价的高性能计算资源。高性能计算集群综合了多台计算机或节点的计算能力,提供了一种经济高效的替代超级计算机的解决方案。集群中的每个用户可以使用不同的编程工具,开发、调试、测试、描述、运行和监看自己的分布式程序。目前,使用集群系统进行的并行计算已在学术界和工业界广泛运用。
网格计算
网格计算关注的是一个集成的计算资源环境。它通过特定的网格软件,将一个庞大的项目分解为无数个相互独立的子任务,然后交由各个计算节点进行计算。在这种系统中,即使某个节点数据出现问题或者突然崩塌,其所承担的计算任务也能够被调度给其他节点继续完成,从而保障整个项目的进程。借助这个模式,普通用户可以获得一种随处可得且可靠、标准且经济的超强的高端计算能力。目前,网格计算广泛的应用于解决各种重大科学应用问题的科学领域。
云计算
云计算是一种基于互联网、定位于服务的超级计算模式。云计算通过虚拟化的关键技术,将各种计算和存储资源进行充分整合和高效利用,构成了典型的数据密集型计算模式。云计算主要提供底层资源,面向大众用户服务,计算支持Web,通用性更强。云计算在医药行业中的应用已成为一种趋势。
殷赋云计算平台是建立在云计算基础上,整合高性能计算集群,专为国内科研院所打造的专业计算平台。该平台将高深的理论计算和分子模拟技术标准化、智能化和精简化,让用户能够快速掌握这一科研利器,提升工作质量和效率。




