- A+
什么是分子动力学?
分子动力学是一套分子模拟方法,该方法主要是依靠计算机来模拟分子、原子体系的运动,是一种多体模拟方法。
在说分子动力学之前,先提一下“量子化学计算”,即基于“第一性原理”(也称“从头算”,即基于量子力学理论的,完全由理论推导而得,不做任何经验值的带入的计算,较为精准,但是速度较慢)。
而很多情况下,对大分子体系的处理可以完全避免使用量子化学计算。分子力学模拟使用古典力学模型(例如谐振子)描述化合物的能量。分子力学模型的所有常数均通过实验数据或第一原理计算结果得到。参数和方程的优化结果称为分子力场。
分子模拟的大致分类
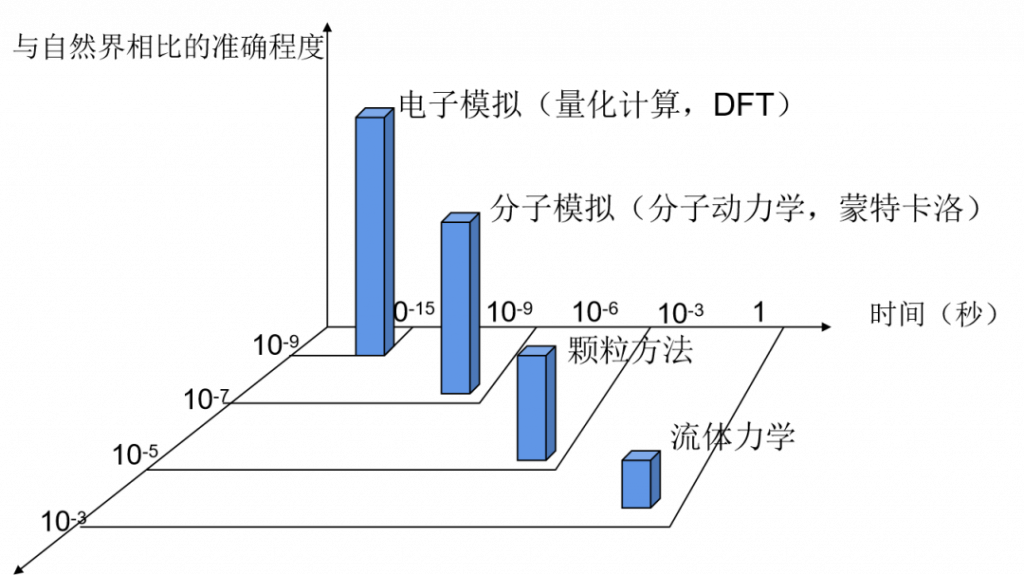
分子动力学通过对分子、原子在一定时间内运动状态的模拟,从而以动态观点考察系统随时间演化的行为。通常,分子、原子的轨迹是通过数值求解牛顿运动方程得到,势能(或其对笛卡尔坐标的一阶偏导数,即力)通常可以由分子间相互作用势能函数、分子力学力场、全始计算给出。对于考虑分子本身的量子效应的体系,往往采用波包近似处理或采用量子力学的费恩曼路径积分表述方式[1]处理。
分子动力学是如何助力高分子研究的?
分子动力学模拟又称MD模拟,自从被学者被提出以来,已经经历了四十年左右的发展历程。MD模拟的研究对象主要针对微观层面,既材料在分子尺度上的性质模拟。
它是一种利用分子在系统中做不规则运动的原理,来解释说明原子或分子在系统中是否相互吸引或相互排斥的方法。[2]
通过粒子间的相互作用力以及空间坐标随时间变化的规律,来解释材料的宏观现象。是一门结合数学,物理和计算机理论的综合技术。
由于体系中的粒子总势在做无规则的运动,可以利用这一特点记录其在确定时刻的坐标和运动轨迹以及势能等的变化,同时用适当的计算方法或统计方法来模拟分析材料的宏观特性。
例如可以计算模拟出材料的力学性能、热学性能甚至化学性能等特性,并结合径向分布函数、均方位移等微观机理对宏观特性进行分析,这种方法对实验方法所测得的宏观性能有一定的补充指导意义。同时在材料模型建立的过程中可以严格地控制变量,规避了实验中可能出现的不确定因素。
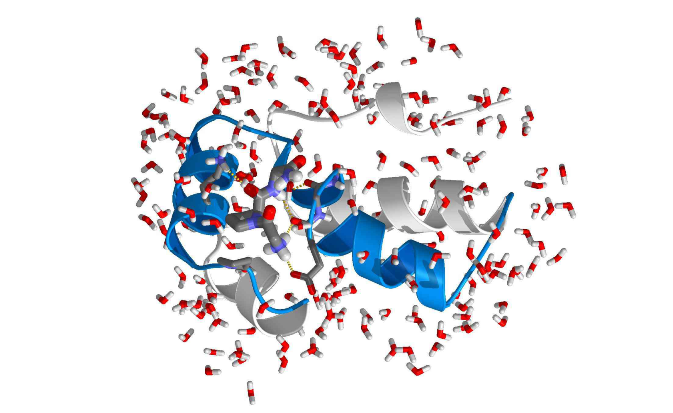
MD模拟还对材料的宏观性能具有一定的预测作用,可以可靠合理的预测实验数据趋势,节省了实验成本和时间,使研究人员可以更有针对地设计试验。
分子动力学研究的具体应用示例

M-N(20-50)的高分子聚合物在温度T = 0.38时的微观结构
高分子聚合物微观动力学性质的分子动力学模拟研究
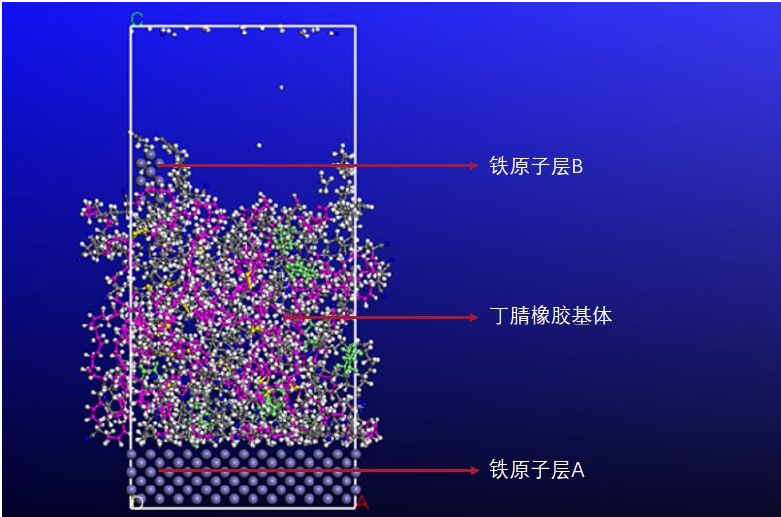
基于分子动力学模拟的丁腈橡胶溶胀及摩擦研究

如今,得力于研究人员的不懈努力,分子动力学的应用越发广泛。作为目前计算大型复杂的分子体系最常用的方法,MD模拟方法也在不断成熟,许多如Material studio、LAMMPS等操作简单运算能力大的软件面世,为科研人员提供许多便利。但同时它也存在占内存过大,计算时间太长等缺点,需要技术的进一步成熟。

结语
随着计算机硬件算力和软件模型的发展,处理大体系、长时间的能力进一步加强,对理论和实验的贡献也在逐步加大。因此,需要加大对分子动力学模拟乃至模拟计算的认识和投入,从而更好地助力科研。




