- A+
近年来,单细胞转录组和表观基因组高通量测序技术的进步为研究界带来了巨大的机遇,扩大了我们对神经科学,发育生物学,免疫学等的认识。基因表达,染色质可及性,DNA甲基化为我们了解单个细胞提供了独特的视角。这些信息共同使我们能够定量推断细胞身份,与基于形态学,表面蛋白和功能的定性定义相比,这是一个巨大的进步。
为了以无偏,全面的方式识别细胞类型和状态,研究者们开发了多种单细胞数据整合方法。在2019年,Joshua Welch课题组开发了LIGER (linked inference of genomic experimental relationships)【1】,该算法基于集成非负矩阵分解 (Integrative Nonnegative Matrix Factorization) 构建,见图1。对于在不同细胞样本上获得的多个单细胞数据集,LIGER通过交替非负最小二乘法 (Alternating Nonnegative Least Squares) 将它们整合,发现与细胞身份相关的特征,同时保留数据集之间的差异。LIGER与Seurat v3和Harmony一起被评为单细胞数据集批次效应校正方面的佼佼者【2】。
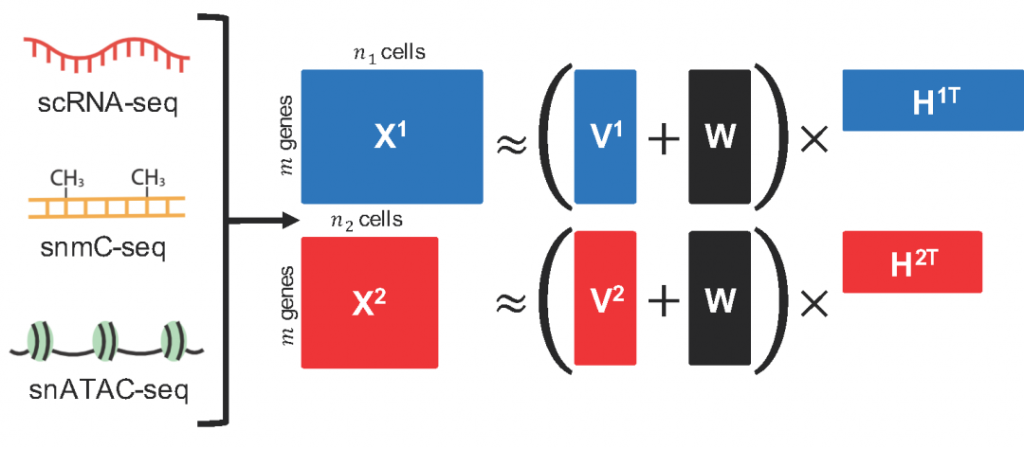
但是,随着可用数据集的大小迅速增加(多达一百万个单细胞),对现有的计算方法形成了挑战。该团队意识到,这些批量学习(Batch Learning)的方法(例如LIGER中运用的ANLS算法)在应对如此庞大的数据集方面效率不高,因为它们需要在整个分析过程中将所有数据存储在计算机内存中。此外,前文提到的这些方法都无法在不从头开始重新计算的情况下整合新数据。
2021年4月19日,美国密西根大学、计算医学和生物信息学系Joshua Welch课题组在Nature Biotechnology杂志上发表文章Iterative single-cell multi-omic integration using online learning,提出将在线学习(online learning)应用于iNMF来改善LIGER。该团队设计的在线iNMF算法 (online iNMF algorithm) 比批量学习具有更快的收敛速度。此外,在计算过程中它并不需要中将所有数据集保留在内存中。相反,它会持续从磁盘加载固定大小的数据 (mini-batch)。由于在每一个时间点只读取和处理一小批数据,因此峰值内存使用量与完整数据集大小是不相关的,这使研究人员在笔记本电脑上分析大规模单细胞数据集成为可能。
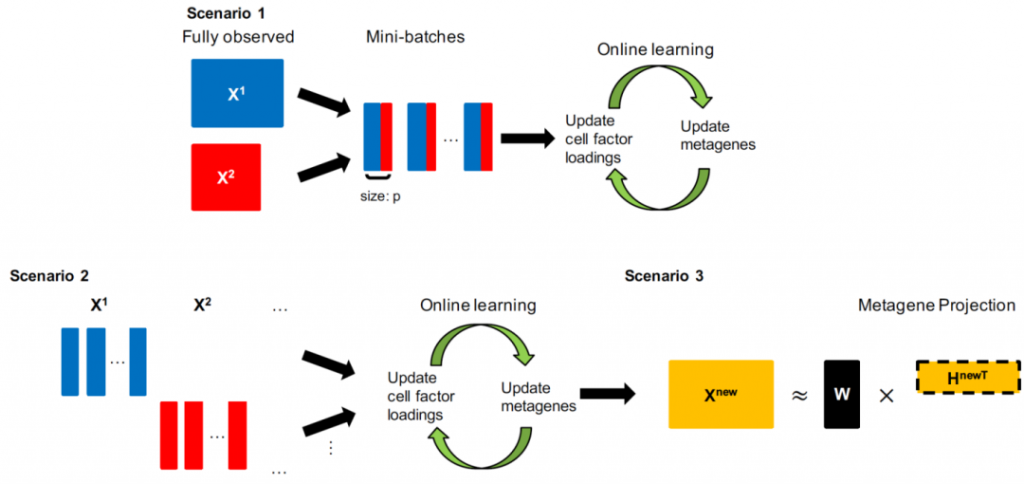
图2. Online iNMF算法的三种应用情境。
在实际运用中 (图2),研究人员可首先按照情境1或情境2使用online iNMF来学习元基因因素,然后使用共享的元基因因素来计算新数据集中细胞的因素负荷。情境3在合并数据方面非常高效,这使用户可以根据指定的参考 (Curated Reference) 对其数据进行矩阵分解,并为新测序数据中的数据集差异提供增强的鲁棒性 (Robustness)。
该团队在多组仿真数据和实例数据上对online iNMF进行了验证。其中实例数据包含了生成于不同物种和组织的单细胞多组学数据:人外周血单核细胞、人胰腺、成年小鼠大脑、小鼠初级运动皮层以及小鼠胚胎。除此之外,该团队还将online iNMF应用于小鼠海马和小鼠下丘脑的大规模空间转录组数据 (Spatial Transcriptomics)。
结果表明,online iNMF算法收敛迅速,并且将峰值内存使用量与数据集大小解耦 (Decouple)。所产生的元基因因素和细胞的因素负荷通过下游分析 (Downstream Analysis) 可得到准确的细胞类型鉴定。该团队希望online iNMF未来能有助于整合来自BRAIN Initiative,Human Body Map和Human Cell Atlas等项目的大型单细胞多组学数据集。
Joshua Welch教授是本文的通讯作者,论文第一作者为博士研究生高超,同课题组成员刘佳麟与April Kriebel为本研究做出重要贡献。加州大学圣地亚哥分校的任兵教授课题组与索尔克生物研究所Joseph Ecker教授课题组提供了数据。




