- A+
用途
处理Amber分子动力学轨迹,方便后续分析。功能包括:轨迹合并、采样、重设时间、截取部分原子、删除水和离子、成像居中和构象叠合。
预备知识
成像居中
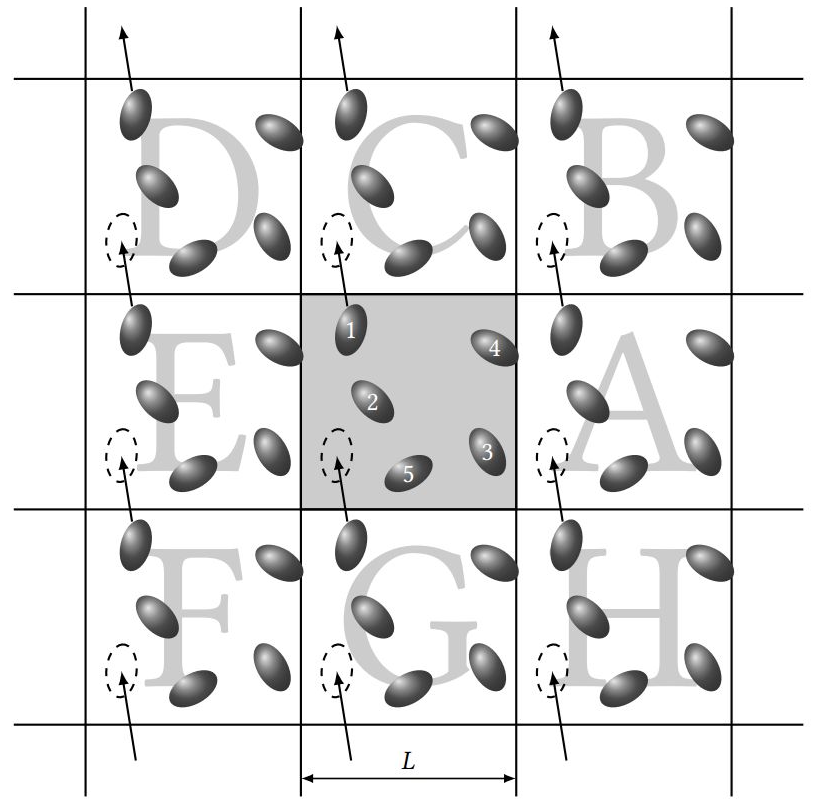
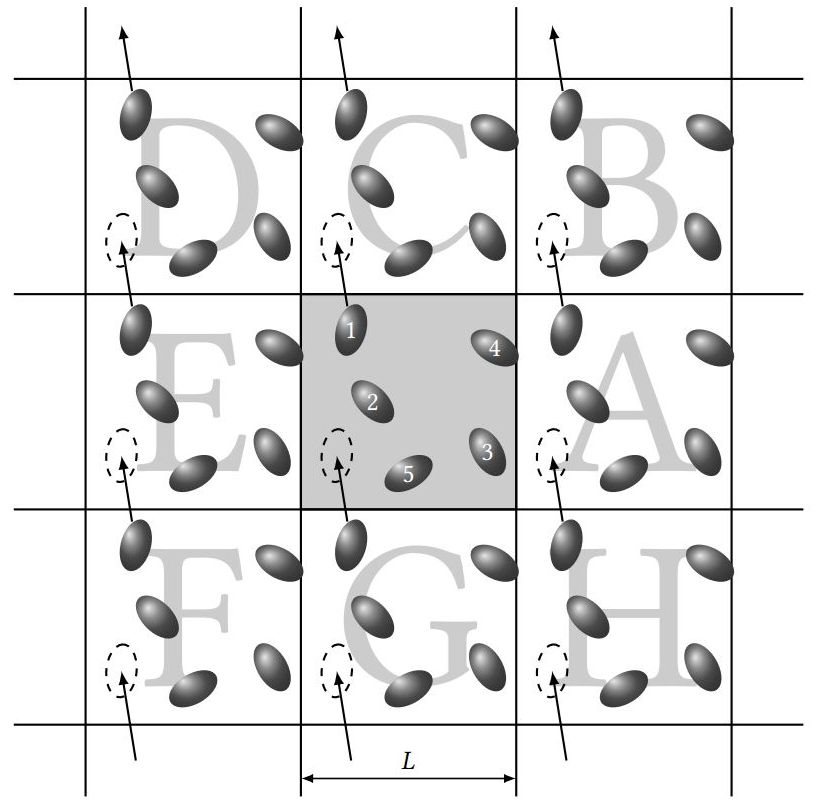
对于包含盒子信息、采用显式溶剂模型和周期性边界条件(PBC)的体系,在长时间的动力学模拟过程中,分子很容易发生“漂移”,导致一部分原子出现在盒子( 元胞)之外,这部分原子又会从盒子另一侧出现(图1),水分子更是容易跑散了,模拟后的结构非常难看,甚至出现横跨盒子的超长的 共价键。为此,需要对轨迹进行 成像居中处理,让镜像原子回到正确的位置,跑偏的分子重新回到盒子中心甚至是坐标原点(图2)。
入口
平台地址:https://cloud.yinfotek.com/
功能入口:左侧菜单栏【计算方案】->【大方案】->【分子动力学】->【处理Amber轨迹】
步骤
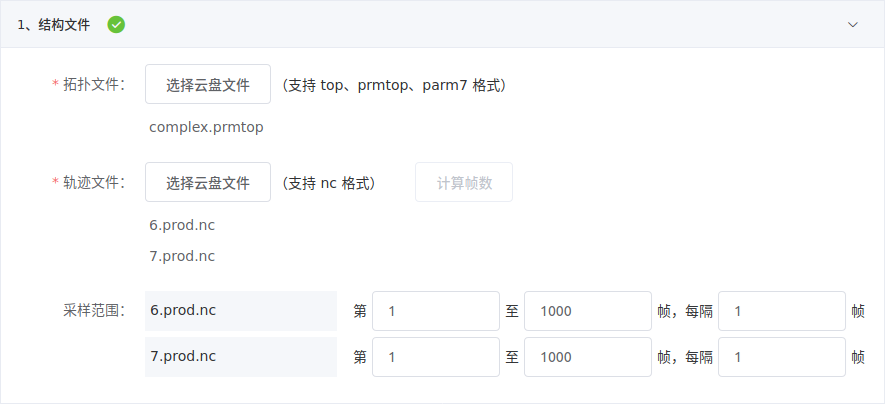
1. 选择结构文件和设定采样范围
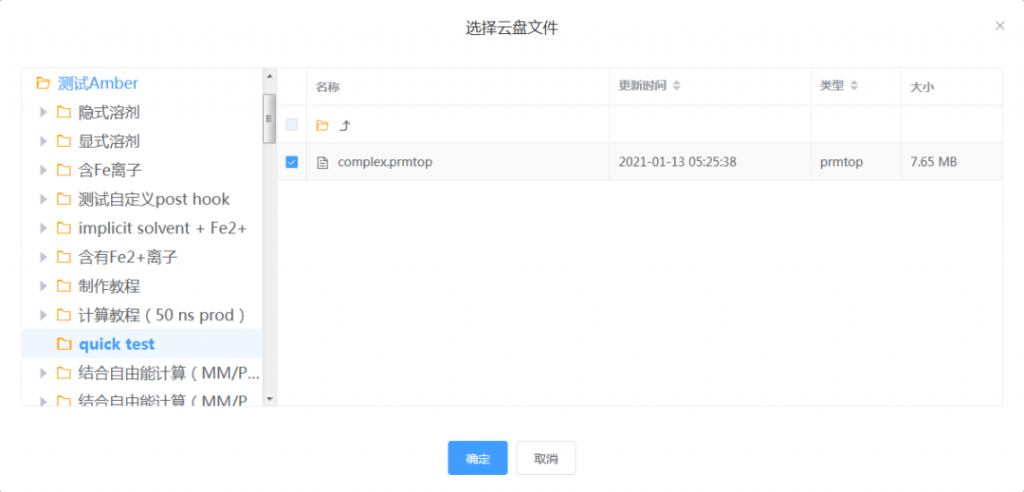
在云盘中,轨迹文件和对应的拓扑文件会存放在相同目录下。
轨迹文件可以多选,但它们必须对应内容相同的拓扑文件,轨迹文件每个原子都与拓扑文件对应。例如,下图所示两个轨迹文件是不同批次的动力学模拟结果,但拓扑文件相同,因此可同时选择。
2. 设置参数
此处提供对轨迹常规处理的功能,大多数情况下采用默认设置即可。
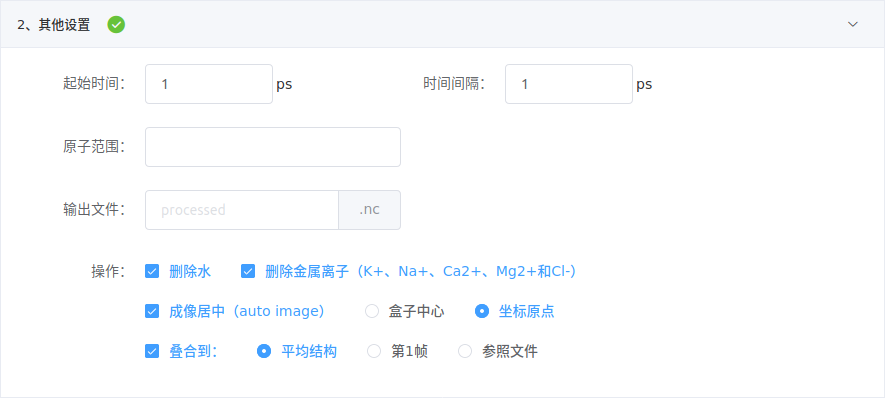
- 模拟时长
对采样得到的轨迹重新设定模拟时间, 起始时间为第一帧的时间起点, 时间间隔为每一帧对应的时长。
一般情况下, 采样间隔等于 时间间隔。用户应在正确理解的前提下根据实际进行设置,否则,不应修改或应咨询客服。

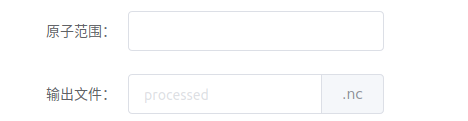
原子范围填写Amber mask(详见【分子动力学模拟(Amber 20)】预备知识),若不填写,则输出全部原子(除非下面操作中勾选删除水或删除金属离子)。输出文件填写文件名。
- 操作处理
- 勾选
删除水或删除金属离子,将从轨迹中删除这些结构;
成像居中会对轨迹每一帧进行居中处理。该项处理只对含有盒子信息(即采用显式溶剂模型和周期性边界条件PBC)的轨迹有效,可居中至盒子中心或坐标原点;
- 勾选
叠合功能,则将每一帧叠合到参照结构上,提供三种参照结构:轨迹所有帧的平均结构、轨迹第1帧或者用户上传的参照文件。
- 勾选

3. 点击【提交】
4. 查看结果
当任务结束后,进入分析页面。可查看到处理后的轨迹信息,从视图查看第一帧构象、最后一帧构象和平均构象,检查是否符合预期。

我的微信公共号
我的微信公招扫一扫



