- A+
写在前面
前述,我们通过《安装 | 比较基因组系列之一 - WGDI 软件安装与配置》一文,带大伙安装和配置了我们 WGDI 软件。接下来,我们直切主题,从实例数据开始,手把手带大家进行比较基因组分析,并做具体结果解读。
比较基因组学的分析工作常常都是从一张点图开始的。一张清晰的点图能反应出来非常多的信息。
初探 WGDI
WGDI 所有的分析,从一个配置文件开始。对于点阵图,我们可以通过运行
wgdi -d ? >> total.conf进而查看配置文件模板
cat total.conf[dotplot]blast = blast file
gff1 = gff1 file
gff2 = gff2 file
lens1 = lens1 file
lens2 = lens2 file
genome1_name = Genome1 name
genome2_name = Genome2 name
multiple = 1
score = 100
evalue = 1e-5
repeat_number = 20
position = order
blast_reverse = false
ancestor_left = none
ancestor_top = none
markersize = 0.5
figsize = 10,10
savefig = savefile(.png,.pdf)下面,逐个参数给大家解读。
blast = blast file,即设置输入的 blast 文件,一般使用物种A蛋白序列全集 比对到 物种B蛋白序列全集,得到blastp结果文件(后续给大家演示)gff1 = gff1 file,即基因位置信息文件,记录每个基因的具体信息,gff 文件使用 Tab键 分割,分别为 chr,id,start,end,stand,order,old_id。其中,order是每条染色体从1开始的排序,old_id 和后面列的信息不读取。
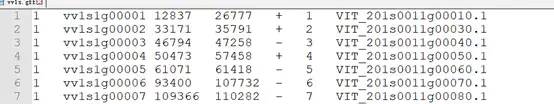
3.lens1 = lens1 file,染色体长度信息,记录每条染色体的长度。三列分别为染色体号,染色体bp长度,染色体基因个数。scaffold或contig 可以等效于chr
其他参数,不重要。主要是blastp结果过滤以及出图参数。
准备示例数据
从上述可以看出,输入数据事实上可以直接从两个文件开始准备。
- 基因组序列文件
- 基因结构注释信息
文件可直接从公开数据库 Phytozome 上下载,此处为v10的
ls -1
total.conf
Vitis_vinifera.genome.fna
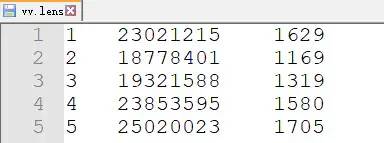
Vitis_vinifera.genome.gff3首先,准备染色体长度信息
perl -0076 -ane '@F=map{s/[>\r\n]//gr}@F;$id=shift @F;print $id,qq{\t},length (join q{},@F),qq{\n} if $id' Vitis_vinifera.genome.fna > Grape.chr.length随后,统计每条染色体上的基因数目(注意,此处统计的是 gene 的数目,如果你的注释信息文件没有 gene 的数目,那么你可能要统计 mRNA 的数目,并注意是否有可变剪切本等)
perl -lane 'next if /^#/;$count{$F[0]}++ if $F[2] eq "gene";END{print join qq{\t},$_,$count{$_} for sort keys %count}' Vitis_vinifera.genome.gff3 > Grape.gene.counts合并两个文件,得到 WGDI 所需的 len 文件
perl -lane 'if($flag){print join qq{\t},$_,$count{$F[0]}}else {$count{$F[0]}=$F[1]}$flag=1 if eof(ARGV)' Grape.gene.counts Grape.chr.length |sort -k1,1V > Grape.len准备 gff 文件
Emmm,perl one-liner 嘛...
# 此处直接使用 mRNA,葡萄注释信息中每个基因只对应了一个mRNAperl -lane 'next unless $F[2] eq "mRNA";/ID=([^;]+)/;push @geneInfo,[$F[0],$1,$F[3],$F[4],$F[6]];END{$preChr="";for(sort {$a->[0] cmp $b->[0] || $a->[2] <=> $b->[2]} @geneInfo){if($preChr ne $_->[0]){$c=0;$preChr=$_->[0]};print join qq{\t},@{$_},++$c}}' Vitis_vinifera.genome.gff3 > Grape.gff准备 BLAST 文件
本例中,我们做的是葡萄内部的,所以只需要提取葡萄的蛋白序列文件,比对到自身即可
gffread Vitis_vinifera.genome.gff3 -g Vitis_vinifera.genome.fna -y Grape.pep.fa
# 清除终止密码子
perl -i -lpe 's/\.$// unless /^>/' Grape.pep.faperl -i -lpe '$_=$1 if /(>\S+)/' Grape.pep.fa比对到自身,推荐 BLASTP,因为准确度也很重要。此处使用 DIAMOND,主要是为了加速
./diamond makedb --in Grape.pep.fa --db Grape.pep.fa
./diamond blastp --db Grape.pep.fa --query Grape.pep.fa --outfmt 6 --evalue 1e-5 --max-target-seqs 20 --out Grape.blastp绘制点阵图
修改配置文件,设置输入的两个文件
[dotplot]
blast = Grape.blastp
gff1 = Grape.gff
gff2 = Grape.gff
lens1 = Grape.len
lens2 = Grape.len
genome1_name = Grape
genome2_name = Grape
multiple = 1
score = 100e
value = 1e-5
repeat_number = 20
position = order
blast_reverse = false
ancestor_left = none
ancestor_top = none
markersize = 0.5
figsize = 10,10
savefig = Grape.dot.png随后运行即可
wgdi -d total.conf
blast = Grape.blastp
gff1 = Grape.gff
gff2 = Grape.gff
lens1 = Grape.len
lens2 = Grape.len
genome1_name = Grape
genome2_name = Grape
multiple = 1
score = 100
evalue = 1e-5
repeat_number = 20
position = order
blast_reverse = false
ancestor_left = none
ancestor_top = none
markersize = 0.5
figsize = 10,10
savefig = Grape.dot.png
findfont: Font family ['Arial'] not found. Falling back to DejaVu Sans.
findfont: Font family ['Arial'] not found. Falling back to DejaVu Sans.查看当前目录,可以看到输出 png 文件Grape.dot.png,直接打开或者下载到本地打开即可
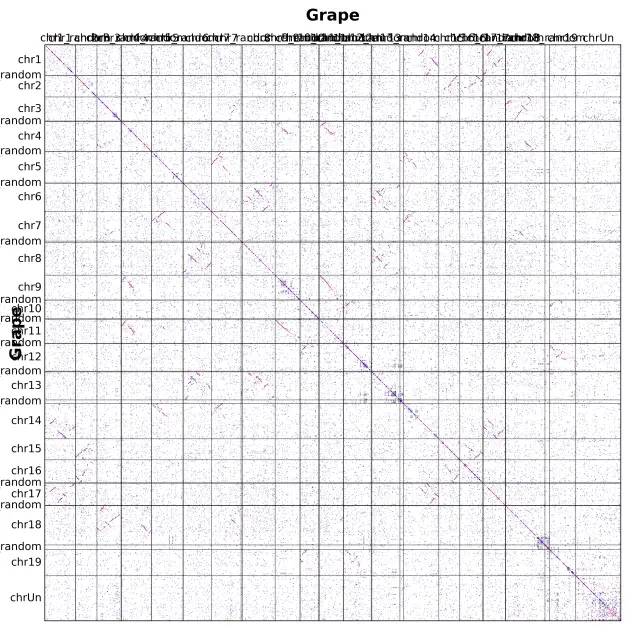
上述,position = order 参数的意思是,基因按照排序位置可视化,我们也可以直接按照具体染色体上的物理位置进行可视化,使用 position = end
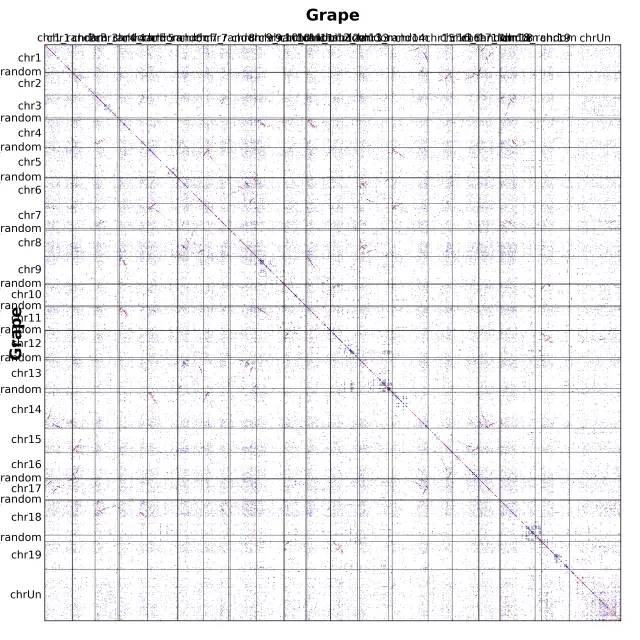
那么为什么要用物理位置可视化?这个与后续 核型分析,祖先染色体重构等相关,继续埋雷,感兴趣的就等后续推送....
与此类似,还有mutiple 参数,表示最优匹配格式,同样与更高维度的比较基因组分析相关。还是埋雷....哈哈
回到主题,任意修改 lens1 和 lens2 的染色体的数量和顺序,可以得到任意染色体间的点图。
比如,我们可以直接去掉 random 染色体碎片
perl -i -lne 'print unless /random/' Grape.len
点图解读
首先,明确 WGDI 点图规则:根据左侧基因组的基因,最优同源(相似度最高)的点为红色,次好的四个基因为蓝色,剩余部分(同源基因有限制个数)为灰色。
- 点图需要横向看和纵向看:这个点图是物种自身比对,所以只需横向看。片段深度应该为 2,但最好同源个数为1,即红色点没有集中分布在某个片段上。常常可以认两个片段很相似,再加上自身。所以,认定为最近发生的加倍为三倍化。除此之外,蓝色或灰色的片段很少有,表明更古老的加倍很不明显。
- 对角线上,本不应该出现片段。自身比自身显然是最好匹配,这些点(WGDI)已经去掉了。所以,其由串联重复造成的,即该基因临近位置的基因相似度很高,为同源基因,打在了对角线附近。可以通过计算这些串联重复的ks值,估算重复片段的爆发时间。
- 最后,事实上,点图是后续跑共线性的基石。score, evalue, repeat_number 判定的同源点的数量是共线性打分矩阵的来源。
写在最后
往往,越是高阶的分析人员,使用的工具却越是简陋。点图,看似简单 和 粗略。但其蕴含的才是真正全面的比较基因组信息。
更多内容,让我们一起期待后续教程。




