- A+
用途
采用MM/PB(GB)SA方法对常规分子动力学轨迹进行结合自由能计算和能量分解,暂不支持计算熵、不支持膜蛋白。
预备知识
MM/PB(GB)SA
计算结合自由能的方法有很多,例如,热力学积分(Thermodynamic Integration,TI)、自由能微扰(Free Energy Perturbation,FEP)、MM/PB(GB)SA、线性相互作用能(Linear Interaction Energy,LIE)。MM/PB(GB)SA方法是精度和速度折衷的方法,广泛应用于受体-配体结合自由能计算。该方法全称 分子力学/泊松-波尔兹曼(广义波恩)表面积(Molecular Mechanics / Poisson Boltzmann (Generalized Born) Surface Area)。顾名思义,该方法将结合自由能拆分成 分子力学项和 溶剂化能分别计算。
其基本原理是:计算两个溶剂化分子在结合(bound)和游离(unbound)状态的结合自由能之差或者比较同一个分子不同的溶剂化构象的自由能。这里以受体蛋白和配体小分子的结合为例,大致讲述其计算原理。
最简单的思路,莫过于按照下图所示的过程直接计算结合自由能,即可分别计算受体、配体以及它们的复合物各自在溶液中的能量,然后算差值:
但在实际计算中,会遇到一个很严重的问题——能量贡献主要来自溶液间相互作用,并且总能量的波动幅度远大于结合能,这样就需要非常长的时间才能收敛。
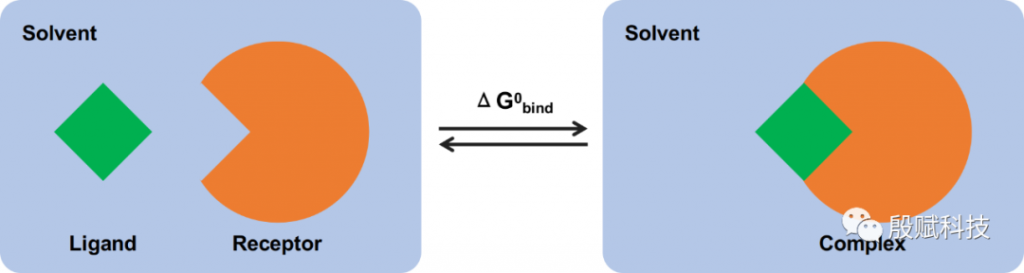
为此,通过下面的热力学循环,“绕个弯”来避免这种糟糕的情况:
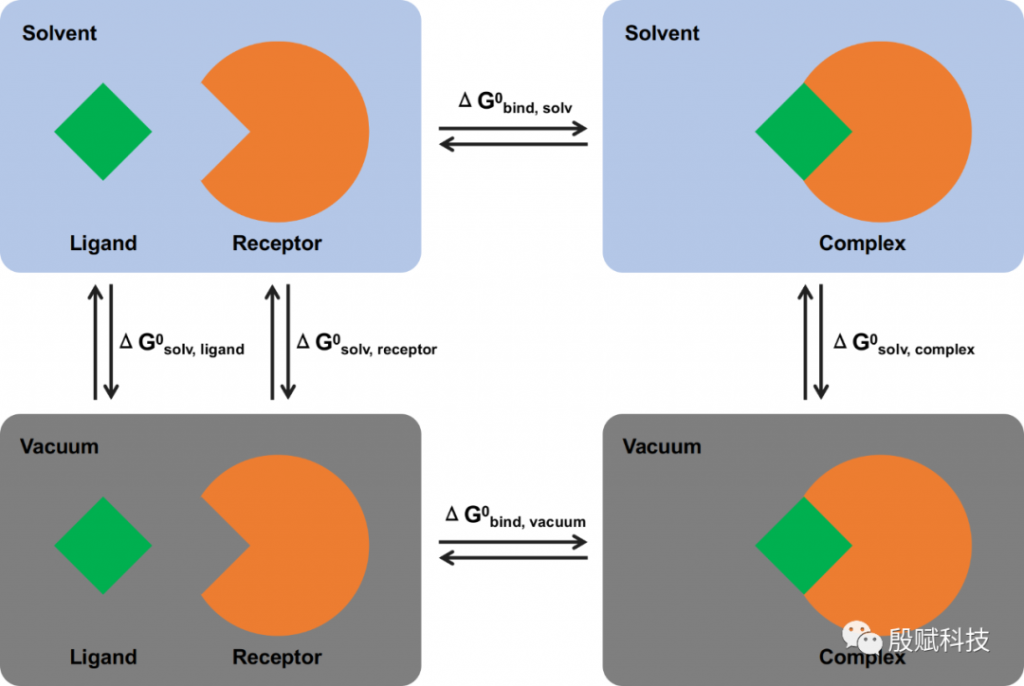
这个方案的含义是,将溶剂中的总结合自由能分拆成分子力学项(真空中的结合自由能)和溶剂化能两部分分别计算。
- 分子力学项
其中, 包含了键、键角和二面角能量, 为非键范德华能量, 为非键静电能量, 为熵贡献,可以采用 简正模分析(normal mode analysis)得到。但实际计算中,通常忽略该部分贡献。因为采用MM/PB(GB)SA方案计算的体系通常为受体-配体结合前后构象变化不大的,这部分贡献可以在计算差值时抵消掉。此外,简正模分析是非常耗时且 误差界限(margin of error)很大、会引入显著的不确定度。
- 溶剂化能
溶剂化能可分成两部分: 极性溶剂化能和 非极性溶剂化能。
对于前者,主要有两种计算方法: Poisson-Boltzmann和 GeneralizedBorn方程,计算较为复杂,这里不展开叙述。对于后者,计算较为简单,它正比于 溶剂可及表面积(solvent-accessible surface area,SASA),它体现了疏水效应,常用的表达式为:
将上述全部公式稍加整理,总结如下:
对于等号右侧的每一项,按照下面公式计算:
(往右移动查看完整公式)
这些对应于MM/PB(GB)SA计算结果中呈现的能量项。
入口
平台地址:https://cloud.yinfotek.com/
功能入口:左侧菜单栏【计算方案】->【大方案】->【分子动力学】->【结合自由能计算(MM/PB(GB)SA)】
步骤
提交任务
1. 从云盘选择 拓扑文件和 轨迹文件
- 拓扑文件必须与轨迹文件原子对应;
- 轨迹文件可多选,但须注意顺序(除非采样范围为全部)。

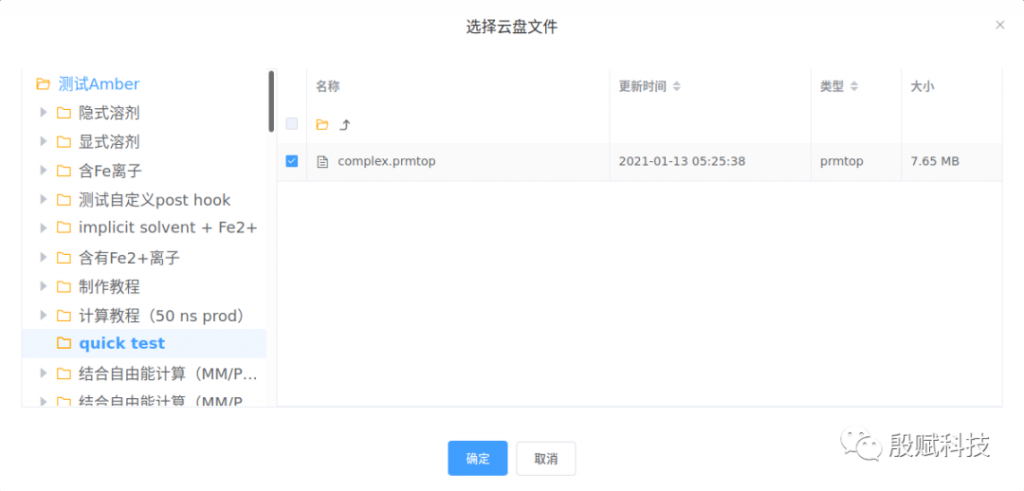
2. 设置采样范围
点击【计算总帧数】获得当前轨迹总帧数,再按需设置采样间隔。
最终采样帧数 = (结束帧号 - 开始帧号 + 1) / 采样间隔。例如,(10000 - 1 + 1) / 50 = 200。
建议对RMSD平稳的那段轨迹进行采样,跨度和间隔也应尽量大。
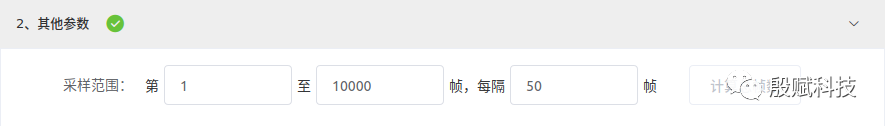
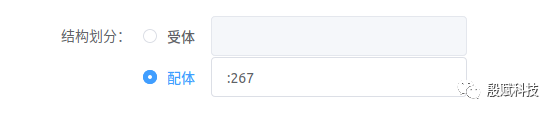
3. 划分 受体和 配体,设置 盐浓度
填写Amber mask(详见【分子动力学模拟(Amber 20)】中的 预备知识),指定受体或配体。
在本例中,受体为1-266号残基,配体为267号残基。因此,有下图所示两种写法。
当体系包含更多成分时,对受体、配体的划分需要特别注意。例如,体系包含蛋白、辅酶、有机小分子、Mg离子(与辅酶配位),想计算有机小分子的结合力,可以这样划分:将蛋白、辅酶、Mg离子归为受体,有机小分子归为配体。
4. 选择盐并设置盐浓度。
虽然MM/GB(PB)SA采用隐式溶剂模型,并未真正添加离子,但不同的盐对应的离子强度不同。

分析结果
1. 查看结合自由能
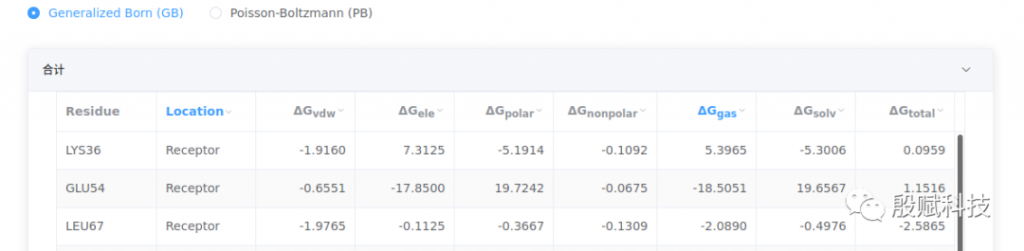
表格给出GB和PB模型的结合自由能数据(详情见 预备知识), mmpbsa.xlsx文件包含了结合自由能数据和能量分解数据。
结合自由能各项能量含义为:
:范德华能量项
:静电能量项
:极性溶剂化能
:非极性溶剂化能
:分子力学项(气相中的能量),
:溶剂化能,
:总结合自由能,
GB和PB模型计算的是溶剂化能部分, 的计算是一样的。结合自由能数值越小,结合力越强。GB模型的 通常比PB模型的值小很多,这意味着两者在溶剂化能计算部分有较大差异。在应用这些数据解析实验现象或探究分子机制时,应注意与实验数据是否冲突,是否能解析得通,选择更符合实际的。

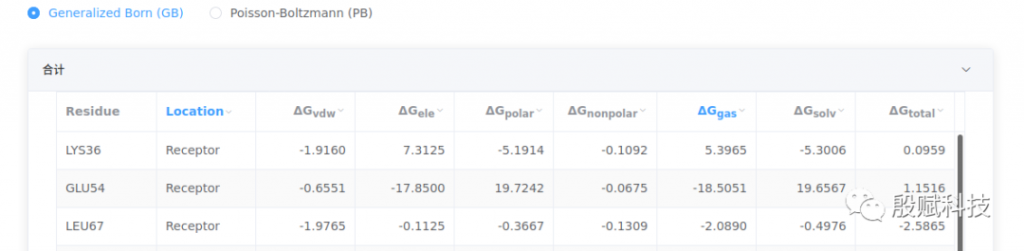
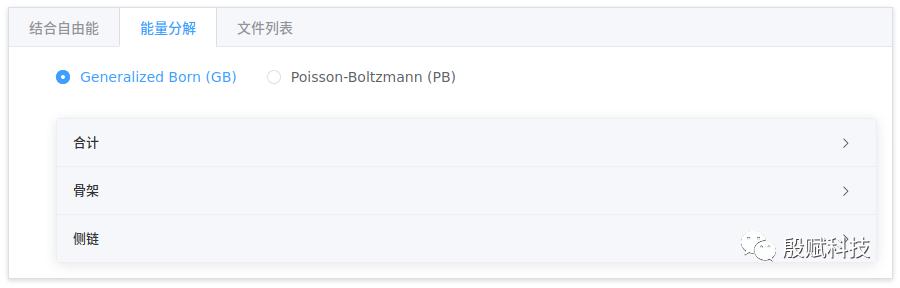
2. 查看能量分解数据
与结合自由能对应,该页也按GB和PB模型分别给出能量分解的数据。
- 每个残基的总能量等于其骨架和侧链的加和。
- 分子动力学模拟中残基编号是从1开始重新排序的,可能与实际编号不一致,请下载 mmpbsa.xlsx文件,修改成实际编号,再进行分析会更为方便。
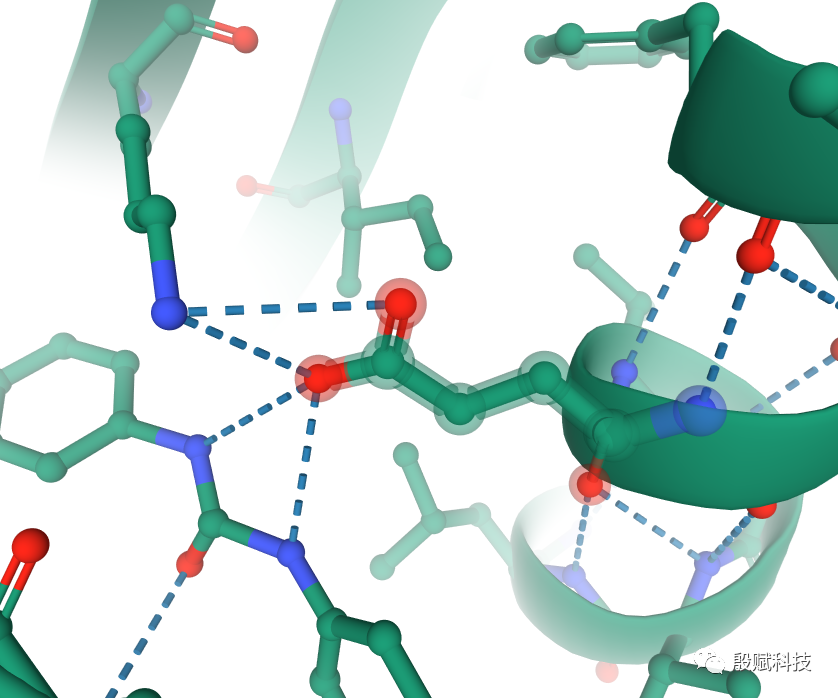
点击表头三角箭头按钮,勾选过滤条件,找到贡献突出的关键残基,再结合3D结构分析,就能精确地了解受体-配体间的相互作用了。
原则上,(正或负)贡献突出的关键残基是 绝对值较大的,但 更能反映出受体与配体之间相互作用大小,与3D结构分析中看到的相互作用力直接对应起来,而 则反映了溶剂化有利于还是不利于结合。
例如,下图分别为GB和PB模型的计算结果。GB结果显示,GLU54的 高达-17.85 kcal/mol,导致其 达到-18.51 kcal/mol。这表明,它与配体分子有强烈的静电力作用,是个关键残基。但 同样很大,致使 达到19.66 kcal/mol,抵消了静电力作用带来的强结合力贡献( 为正数)。但这个残基并非是多余的,从3D结构中可以清楚看到,它与配体和邻近的赖氨酸形成强烈的盐桥作用。
再查看PB给出的 ,尽管仍然为正数,但比GB小很多, 已为负数(-5.77 kcal/mol)。由此可知,该残基确实是关键残基,PB计算值更符合结构分析结果,应采用PB的数据分析本体系。
实际上,侯廷军等人在2012年学术年会的一篇评估研究报告指出,MM/PBSA的预测结果更接近实验数值,而MM/GBSA则对相对结合自由能预测性能更好。对MM/PB(GB)SA方法计算的结合自由能,应结合实际进行取舍。
另外,需要注意的是,PB模型未实现对非极性溶剂化能( )的分解,因此该项数值总是0,但通常绝对值很小,不影响分析。