- A+
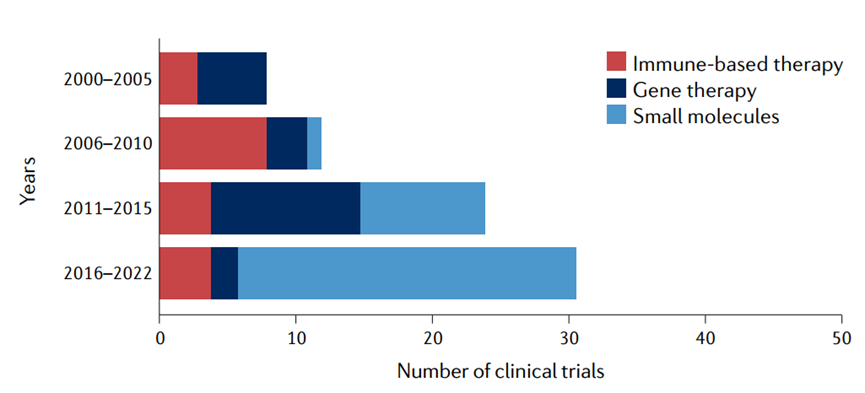
早在10年前,人们就试图通过恢复p53蛋白的功能来治疗肿瘤,但基于p53治疗方案的药物至今也没能被批准上市的。很大程度上,p53作为转录因子不具备典型的可药靶点的特征。随着技术的进步,靶向p53的治疗策略又开始出现新转机,让p53免于被负调节子调控或恢复p53突变体的活性的药物小分子引起了人们极大的兴趣,同时基因疗法和基于p53的免疫疗法也逐渐兴起。靶向p53仍然面临诸多问题,这篇Nature Reviews Drug Discovery上发表的综述文章,重新审视了靶向p53在肿瘤中的努力和困境。

p53肿瘤抑制蛋白是抑制肿瘤发生和发展的主要关卡,它作为转录因子可以激活相邻基因的表达或者在增强子的作用下激活远端基因。通过E3泛素连接酶MDM2的调控,p53在正常细胞中维持较低水平,MDM2是p53的主要抑制子,还有一个抑制蛋白是MDM4。TP53基因的突变使得其编码的p53蛋白失去肿瘤抑制活性,因此10年前人们试图通过恢复p53蛋白的活性来抑制肿瘤,然而一直没有大的进展,很少有药物能够从初始走向临床试验阶段,而且至今也
没有药物获得FDA批准上市。(除非明确强调,本文TP53表示编码p53的基因,而p53则指代蛋白)
TP53基因突变主要的后果是抑制肿瘤功能的丧失,因此需要治疗策略可以恢复p53的功能,然而大部分小分子的功能是抑制过表达蛋白的活性,因此p53被认为是不可药靶点。然而,最近的一些基于p53的治疗方法的兴起,让靶向p53的策略重回人们视线。其中一些方法是早期治疗尝试的延续,改进的方法纳入了新的知识和对作用机制和药物递送方式的更深入的理解。其他一些获得进展的方法是在药物设计领域,如针对性地降解任何选择的蛋白质的新策略。能够保护p53免受其负调控因子或恢复p53突变体功能的小分子越来越引起人们的兴趣,并且针对特定突变体的精准药物设计也已经取得了进展。
与此同时,人们对基因治疗策略重新产生了兴趣,这些策略在上世纪90年代得到了广泛的支持。从二十一世纪最初几年里的被冷落,基因治疗的策略如今重新开始复苏,包括p53领域。同样,属于癌症的免疫治疗方法也在复兴。免疫治疗已经彻底改变了以前几种高度致命的癌症的治疗,这也激发了人们对招募免疫系统来识别和攻击TP53突变癌细胞的兴趣。越来越多的研究表明,癌细胞中p53功能的丧失会对肿瘤免疫微环境(TIME)产生细胞非自主性的影响,使癌细胞能够更好地逃避免疫攻击。因此,在这些癌细胞中恢复p53的功能有望使它们对免疫检查点抑制等方案敏感,从而提高探索相关药物组合的可能。
尽管基于p53的治疗在过去10年中已经在一些优秀的综述中得到了讨论,但新方法的引入和正在进行的越来越多的临床试验需要不断地重新评估当前的认知。本综述试图重新评估利用恢复或增强癌细胞中野生型p53活性的小分子靶向p53功能失调的癌症的成果,并对基于p53的免疫治疗策略和基于p53的基因治疗进行概述,还讨论了将此类治疗引入治疗所面临的挑战和困境。

p53,癌症中极具吸引力的靶点
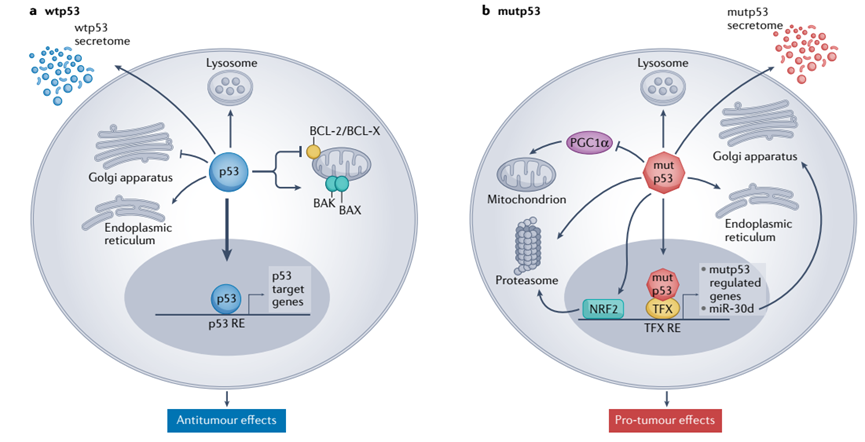
自1979年p53被发现以来,人们发现了p53与癌症的密切关系。p53作为转录因子,可以调控促进细胞细胞周期停滞、凋亡、DNA修复相关基因的表达。事实上,p53影响着所有的细胞器,如线粒体、溶酶体和内质网,如图1所示。突变的TP53会导致p53蛋白的失活,且主要是错义突变导致的单个氨基酸的替换。p53突变体可达数百之多,一些出现频率高的突变被称为热点突变。 和癌症相关的p53突变主要分为两类,一种是DNA接触氨基酸的突变,这种突变对结构影响非常小;另一种是构象突变,可以导致结构较大的改变,甚至错误的折叠。无论是突变体p53或者野生型p53都可以调节肿瘤微环境,达到抑制或者促进肿瘤的效果。例如,p53可以通过控制外泌体携带的microRNA的组成和分泌细胞因子的模式来抑制肿瘤的进展,从而分别维持肿瘤相关神经的分化状态和抑制中性粒细胞浸润。相反,p53突变体可以通过调节外泌体的含量来支持肿瘤的进展,从而导致巨噬细胞重编程为M2状态,从而产生更有利的肿瘤微环境。
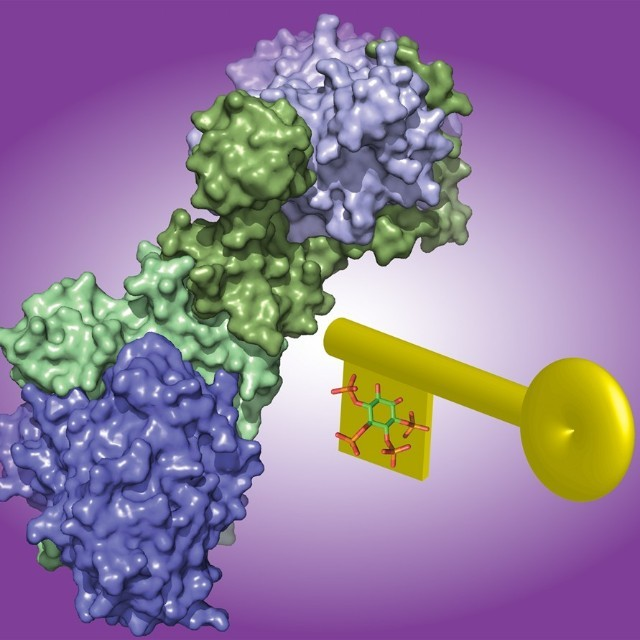
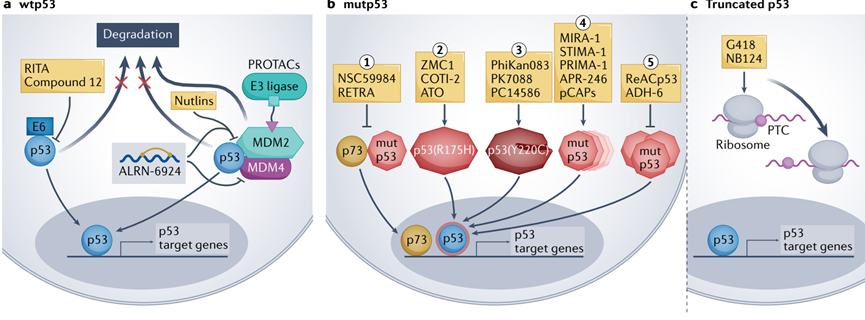
野生型p53对癌细胞和肿瘤微环境(TME)的多种有害作用,如诱导细胞死亡或复制性衰老和促进肿瘤限制性免疫TME,可能解释了为什么TP53在人类癌症中如此频繁地发生突变 (图1 )。这也使得恢复癌细胞中的p53活性是非常有前途的肿瘤治疗策略。尽管仍有许多障碍有待克服,但我们对靶向p53的理解和能力正在逐渐提高,为最终影响许多患者的生活带来了转机。靶向p53的小分子对能够激活p53活性的小分子的追求始于几十年前,此后一直在稳步增强(图2 )。重要的是,具有不同TP53状态的肿瘤需要非常不同的小分子策略。因此,对于存在TP53错义突变的肿瘤,小分子药物的研发主要集中于将突变体恢复到和野生型构象和活性相当的状态。相反,对于具有p53活性的癌症,主要的方法是发现小分子,使p53免受其负调控因子的抑制,最明显的是MDM2,从而释放出完整的p53活性。
靶向TP53错义突变的癌症
如前所述,TP53错义突变在人类癌症中异常常见,约占所有TP53改变的70 %, 这些构象突变被认为是有希望的小分子靶标,通过小分子恢复到正确的折叠和功能。第一个具有p53突变体激活能力的化合物于1999年被发现。CP31398,由Pfizer通过合成化合物库筛选鉴定,恢复了p53的转录活性,并减少了体内肿瘤的生长。从机制上推测,具有迈克尔受体特性的CP31398通过抑制p53的泛素化来稳定p53的野生型构象并阻止其降解。后来该药物被发现严重的副作用, 因为它影响了DNA和p53非依赖性上调促凋亡蛋白BAX的结合。因此,CP31398没有进入临床开发。尽管如此,这种原型药物标志着靶向p53的小分子时代的开始
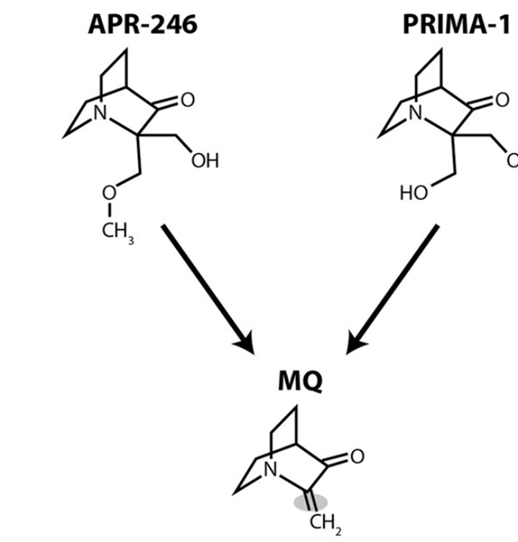
2002年,化学药物筛选发现PRIMA-1可以与p53突变体结合并且恢复p53功能,能促进表达 R273H突变蛋白的Saos-2细胞凋亡,并抑制这些细胞在体内形成肿瘤。药理学上,PRIMA-1实际上是一种前药;它的降解产物MQ与p53突变体核心结构域中半胱氨酸残基的巯基共价反应,恢复野生型p53的构象。此外,MQ可通过直接与谷胱甘肽中的半胱氨酸结合并通过抑制硫氧还蛋白还原酶来改变细胞氧化还原平衡,从而通过破坏维持细胞活力所需的关键稳态机制促进癌细胞死亡。此外,PRIMA-1还通过诱导BAX依赖的细胞色素c从线粒体释放,从而驱动caspase激活,促进细胞凋亡。
与PRIMA-1一样,MIRA-1和STIMA-1也具有迈克尔受体活性,并可能修饰p53蛋白中的半胱氨酸,以稳定野生型构象和阻止p53突变体解折叠。虽然这两种化合物在体外和体内都显示出p53依赖的效应,但由于溶解性问题(STIMA-1)和对正常细胞的毒性( MIRA-1),这两种化合物都没有进入临床试验(图3 )。
在另一种方法中,使用噬菌体展示选择开发了一组p53突变体激活小肽( pCAP ),并在体外和体内应用于含有TP53错义突变的癌细胞时显示出类似野生型p53的效果。机制研究表明pCAP优先与野生型p53构象结合;当p53突变体分子呈短暂的类野生型构象时,肽结合并稳定它,使p53群体的动态平衡逐渐向该方向移动。
尽管如此,仅有少数报道的p53突变体再激活小分子已进入临床试验(表1 )。第一个达到临床试验的是PRIMA-1的甲基化衍生物,也称为APR - 246。在体外和临床前研究中,APR-246表现出更好的活性。PRIMA-1,在急性髓系白血病( AML )细胞系和患者原代细胞中凋亡效应增加,APR-246促进了小细胞肺癌(SCLC)细胞系的凋亡,并延缓了注射此类细胞的小鼠的肿瘤生长。但是APR-246对具有热点突变Y220C的细胞没有促进凋亡的作用,表明该化合物并不是对所有突变体有效。
除了作为单药治疗时的效果外,APR-246与体外化疗以及肺癌和卵巢癌小鼠异种移植模型联合使用时也增加了癌细胞死亡。重要的是,在TP53错义突变患者的原代卵巢癌细胞中,这种协同作用比那些携带非错义突变或者正常p53的患者更有效。
目前正在进行APR-246的临床试验。APR-246与FDA批准的首个治疗骨髓增生异常综合征( MDS )的药物阿扎胞苷联用,在携带p53突变的MDS或AML患者中,Ⅰ和Ⅱ期试验显示出显著疗效。该组合具有很好的耐受性,并显示了一个类似的安全窗口。一些携带TP53截短突变的患者也从APR-246治疗中获得临床获益,表明APR-246的p53非依赖性效应也有助于临床结果。
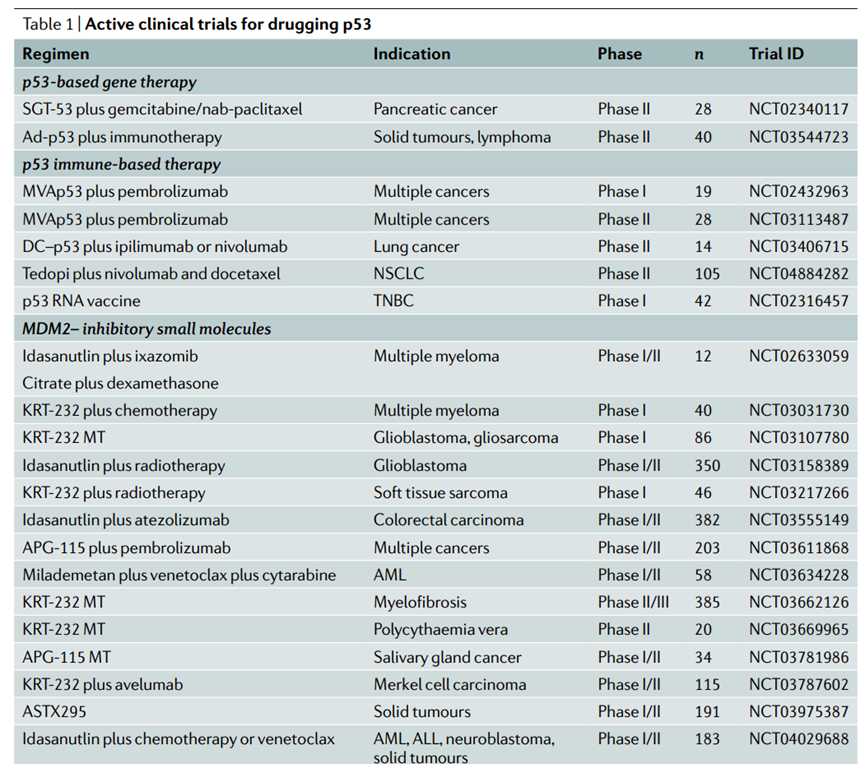
2020年1月,FDA批准APR-246用于MDS治疗,并启动了联合治疗的Ⅲ期多中心随机试验( NCT03745716 )。然而,尽管该试验结果显示联合治疗组的完全缓解率为33 %,而单用阿扎胞苷组的完全缓解率仅为22 %,但未达到统计学意义。最近,一项评估APR-246联合阿扎胞苷作为TP53突变的MDS和AML患者移植后管理治疗的Ⅱ期试验公布了令人鼓舞的结果。与先前的试验相比,移植后1年无复发生存率( RFS )约为30 %,中位总生存期( OS )为5-8个月,与APR-246和阿扎胞苷联合治疗相比,RFS为58 %,中位OS为19.3个月。以APR-246为单一疗法或联合疗法,以及口服下一代化合物APR-548 (NCT04638309),以血液癌症和实体瘤为目标正在进行或计划进行更多的试验。
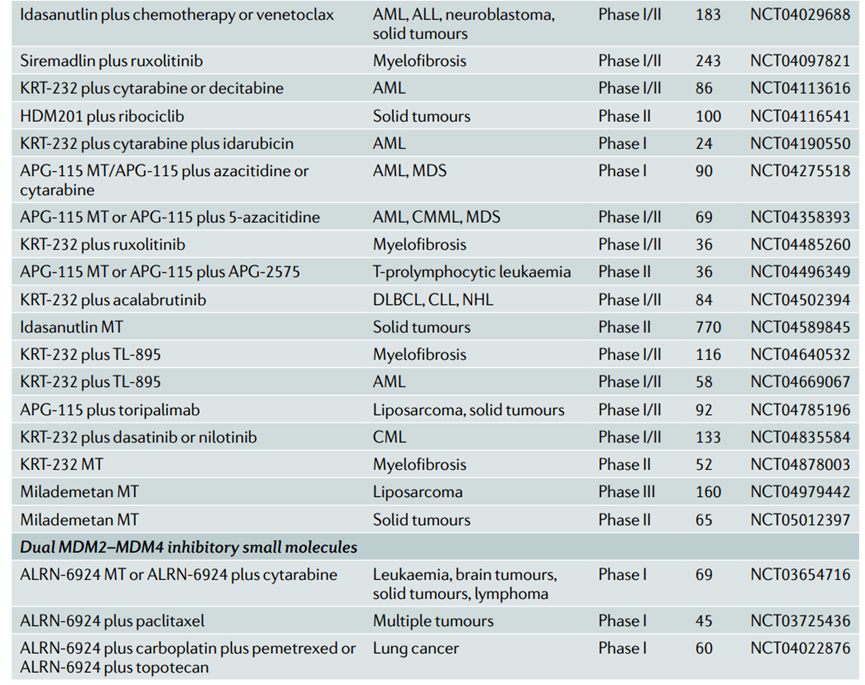
破坏p53突变体与p73之间的相互作用并释放其肿瘤抑制活性的小分子也有报道,p73和p53共享多个转录靶标,但是能被突变体p53抑制活性。将p73从p53突变体中释放出来的分子使其能够引起野生型p53同样效应,包括生长抑制和癌细胞死亡。
一个例子是在一个化学库筛选中被发现了转录报告子活性的重新激活(RETRA),并化合物显示在体外促进细胞凋亡和体内肿瘤消退。另一个小分子NSC59984也可以破坏p53突变体和p73复合物。有趣的是,NSC59984最近被证明在结直肠癌细胞系和小鼠移植瘤中诱导p53突变体降解,导致癌细胞死亡。在机制上,NSC59984被发现在活性氧( reactive oxygen species,ROS )存在的情况下促进ERK和MDM2磷酸化,导致MDM2-p53突变体结合,p53突变体泛素化和然后被蛋白酶体降解。
错义p53突变体蛋白可以形成分子聚集体,并可能通过隔离p73和p63促进增强突变(GOF)效应。这推动了一项理性的设计研究 ReACp53,一种抑制p53突变体聚集的肽。ReACp53恢复了p53的野生型构象和核定位,使p53依赖的基因转录,并在体外促进癌细胞凋亡和细胞周期阻滞,以及在卵巢癌模型中的体内肿瘤抑制。
随后,通过筛选之前被证明可以抑制阿尔茨海默病相关淀粉样蛋白形成的寡吡啶基酰胺库,三联吡啶酰胺ADH-6被鉴定为p53突变体聚集的抑制剂,可以增强细胞死亡和抑制肿瘤生长,对表达p53突变体的癌细胞具有高选择性,对健康组织或正常p53的细胞没有毒性,并且比ReACp53更有效。但这些药物分子都没能进入临床。

虽然上述化合物是相对广谱的,每一个针对不同的p53错义突变体,旨在具体针对单一突变体或一组不同的类似突变体的方法也被重视。对p53 (Y220C)突变蛋白的高分辨率晶体结构分析发现,在突变位点附近存在一个可进入的缝隙。使用基于结构的虚拟筛选,小分子PhiKan083被发现结合这个空隙中,并在热力学上稳定p53 (Y220C),将其转移到一个类似野生型p53的状态。随后对合成的片段库进行筛选,得到PK7088,其与Phi Kan083一样,结合p53 (Y220C)空腔并稳定其正确折叠。PK7088诱导含有Y220C突变胃癌和肝母细胞瘤细胞凋亡并降低细胞活力,并与野生型p53激活药nutlin-3合作反式激活p53靶基因。
这些效应对于携带Y220C突变的癌细胞是特异性的。PMV制药公司也采用了类似的方法,产生了p53 (Y220C)特异性小分子PC14586。PC14586,目前正在进行I/II期临床试验(NCT04585750),并在最近的ASCO年会上提出了令人鼓舞的初步结果。总体而言,p53 (Y220C)特异性药物如PC14586的成功令人鼓舞。然而,Y220C突变并不十分丰富,其独特的结构使这类药物的开发与其他p53突变体并不共享。因此,将类似的基于结构的方法应用于其他p53突变体可能更具挑战性。
P53 (R175H)属于p53突变体的一类,其中DNA结合结构域的构象严重扭曲,因此被称为结构或构象突变体。具体来说,p53 (R175H)突变导致锌离子结合受损,引起p53蛋白的错误折叠和失活。
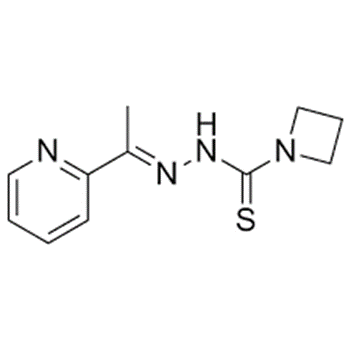
NSC319726 (也称为ZMC1),一种对锌具有高亲和力的金属离子螯合剂,被确定为p53 (R175H)靶向药物,同样基于计算机筛选得到的药物分子,NSC319726在体外以高度特异于p53 ( R175H )的方式促进p53依赖的细胞凋亡和体内肿瘤消退。非转化细胞和含有野生型p53的癌细胞不受影响,而表达其他p53突变体如p53 (R273H)和p53 (R248Q)的细胞则丧失了p53参与序列特异性的能力,在不引起蛋白( DNA接触突变体)整体结构严重扭曲的情况下,DNA结合仅受到轻微影响。
此外,NSC319726抑制了移植有p53 (R175H)突变的人癌细胞异种移植的小鼠以及携带p53 (R172H)突变的敲入小鼠体内肿瘤的生长。同属缩氨基硫脲家族的ZMC2和ZMC3也被发现在体外促进p53 (R175H)的野生型构象。在整合的计算平台中鉴定的第三代缩氨基硫脲COTI2对p53突变的癌细胞系表现出优先选择性,但在p53野生型细胞中也具有一定的活性。事实上,随后在头颈鳞状细胞癌(HNSCC)细胞、体外和原位小鼠模型中的研究证实,COTI2具有p53依赖性和p53非依赖性两种作用,目前COTI2已经进入临床一期试验。
最近,Chen等通过生物信息学分析发现,FDA批准的治疗急性早幼粒细胞白血病的药物三氧化二砷( ATO ;三氧二砷)可以挽救结构p53突变体的功能,而对DNA接触突变体的作用有限。有趣的是,尽管ATO恢复了广泛的p53突变体的正确折叠,但只有一部分p53突变体恢复了野生型p53样的转录活性。目前正在进行临床一期试验,用地西他滨和ATO联合治疗TP53突变的AML和MDS患者(NCT03855371)。ATO作为突变体p53再激活剂的"再发现"增加了一个有趣的可能性,那就是其他的FDA批准的小分子化合物,包括一些用于治疗癌症以外的疾病的化合物,也可能具有不受重视的野生型p53再激活能力。靶向野生型p53MDM2和MDM4抑制剂
在保留野生型p53表达的肿瘤中,以p53为靶点的治疗方法最广泛的是抑制p53的降解。目前研究最透彻的p53降解机制是通过E3泛素连接酶泛素化MDM2,导致p53的蛋白酶体降解。重要的是,在许多保留有p53的癌症类型中观察到的MDM2扩增。MDM2介导的泛素化和降解依赖于其与p53的直接结合,这促使了寻找抑制MDM2-p53结合的小分子作为稳定p53的手段。
第一种此类抑制剂是由Vassilev等人通过合成化学库筛选确定的顺式咪唑啉nutlins。它在野生型p53癌细胞中引起p53激活,而在p53突变体细胞中没有作用。nutlins 衍生物RG7112是第一个临床试验的MDM2抑制剂。在难治复发AML和慢性粒细胞白血病( CML )患者中,RG7112触发野生型p53激活,包括p53蛋白稳定,以及许多p53靶基因的表达升高,如CDKN1A和BBC3。
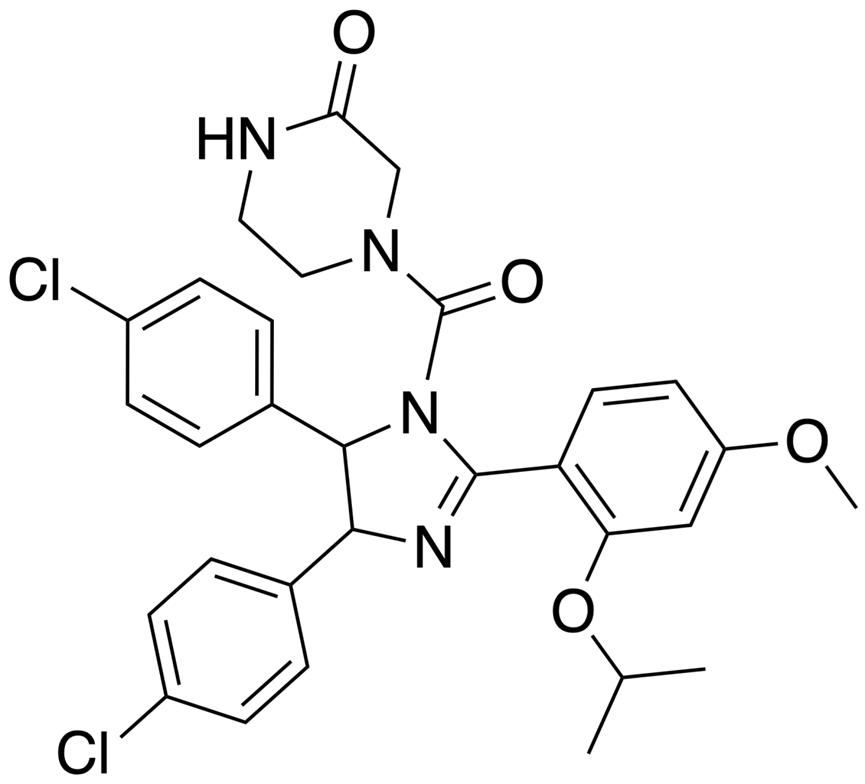
令人欣慰的是,在许多患者中观察到了抗白血病活性。出乎意料的是,在少数没有TP53突变的患者中也发现了临床活性,这表明RG7112也可能具有p53非依赖的活性,如通过缺氧诱导因子1α (HIF1α)抑制血管生成。然而,需要高剂量的RG7112才能达到疗效,这会导致胃肠道不耐受和抑制血小板生成的不良反应。同样,在脂肪肉瘤患者中,高剂量的RG7112与血小板减少和中性粒细胞减少相关。
除了nutlins 衍生物,许多其他干扰MDM2-p53结合的分子已经或正在被开发和测试。例如,口服生物可利用的强效MDM2抑制剂APG-115在AML83的临床前模型中显示出强大的抗肿瘤作用,并使胃癌移植瘤对放疗敏感。APG-115目前正在几个临床试验中进行评估(NCT02935907,NCT03611868,NCT04785196,NCT03781986),单药治疗或联合化疗或免疫检查点抑制(表1)。AMG 232是另一种口服生物可利用的MDM2抑制剂,显示在骨肉瘤细胞中促进野生型p53功能和肿瘤消退,该药物也在进行多项临床试验。
虽然MDM2抑制是一种有吸引力的策略,但新的MDM2抑制剂是否对正常组织的危害较小仍有待观察。p53在几乎所有的正常组织中都有表达,特别是在它们的增殖区。它不是癌症特异性的目标,努力生产一种绝对无不良反应的MDM2抑制剂很可能不是一个现实的目标,除非能够找到一种MDM2抑制剂与癌症特异性模式相结合的较低、耐受性好的组合,或者将MDM2抑制剂选择性地递送到癌细胞。
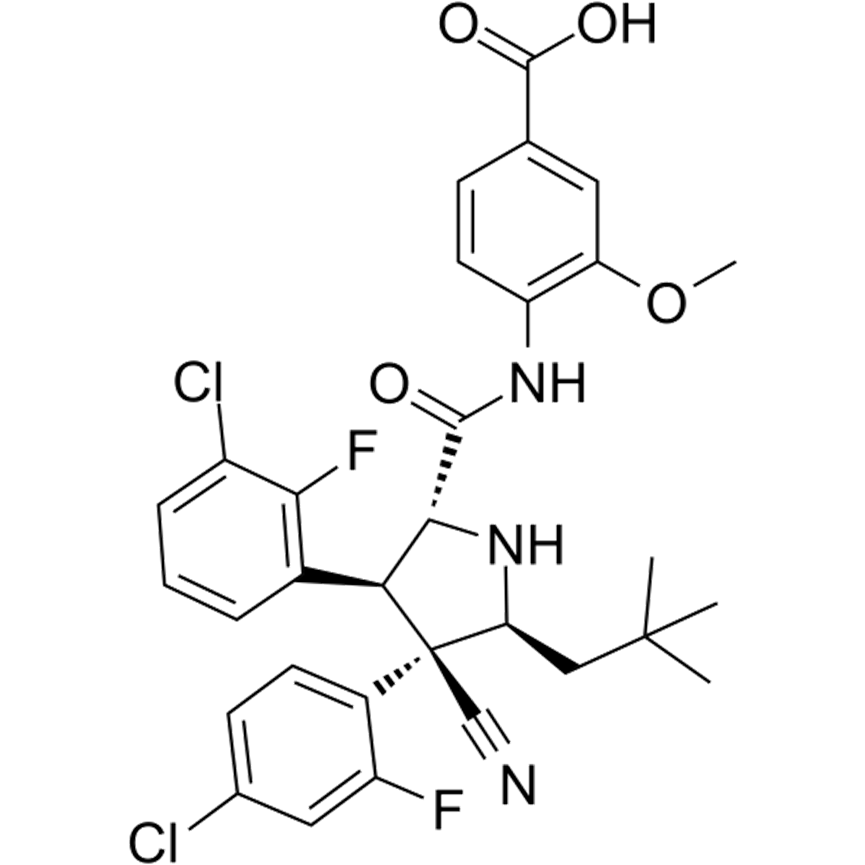
MDM2相关蛋白MDM4也是p53的重要负调控因子。与MDM2不同,MDM4不具有内在的E3泛素连接酶活性。然而,它可以促进MDM2的E3连接酶活性,也可以直接与p53结合并抑制其转录活性。与MDM2一样,MDM4在许多癌症中扩增,使其成为有吸引力的治疗靶点。值得注意的是,血液系统恶性肿瘤,包括AML和骨髓纤维化,大多保留野生型p53,通常伴随着MDM2或MDM4的高表达。MDM2、MDM4在白血病干细胞中高表达,使得MDM4抑制剂特别有希望用于白血病治疗。近年来,短肽作为小分子药物的替代品应运而生。Bernal等通过化学碳氢化合物拼接,开发了一种能够阻断MDM2和MDM4与p53相互作用的短肽( SAH-p53-8 )。然而,随后的体外实验发现SAH-p53-8可能具有p53非依赖的细胞毒性,引起了人们对其临床应用的担忧。
后来,额外的靶向MDM2和MDM4的双特异性短肽被引入,ALRN-6924对含有野生型p53的多种乳腺癌细胞系表现出较高的疗效,但是p53突变型细胞则对其耐受。在AML细胞系中也发现了类似的p53选择性,其效果优于idasanutlin,而p53敲低则取消了这些效果。临床I试用ALRN- 6924于2016年启动,在实体瘤和淋巴瘤中发现了抗肿瘤活性,副作用可以容忍。当前,有3项关于ALRN-6924的临床试验正在进行中。
常规的小分子抑制剂MDM2和MDM4以化学计量的方式工作,通常需要相对较高的剂量。相比之下,蛋白水解靶向嵌合体(PROTACT)和分子胶复合物具有催化功能,即使在低剂量下也能发挥有效作用。PROTACT是小的异双功能分子,通过它们的一个手臂与E3泛素连接酶结合,通过它们的另一个手臂与目标蛋白结合。蛋白裂解靶向嵌合体通过将目标蛋白正确定位在E3连接酶附近的物理位置,促进了前者的泛素化和随后的蛋白酶体降解。作为一种E3连接酶,MDM2也被用于通过PROTAC介导的招募来降解其他致癌蛋白。特别是,基于这样的知识,Nutlin紧紧地绑定到MDM2,基于Nutlin蛋白裂解靶向嵌合体已研制成功。
当应用于携带p53的癌细胞时,这样的蛋白裂解靶向嵌合体就像一把双刃剑,例如一个基于核仁素的PROTAC靶向转录和表观遗传调控因子BRD4:在促进BRD4降解的同时,它也抑制MDM2,导致p53激活。此外,PROTAC方法学也被用于靶向MDM2本身。一个这样的例子是 MD-224在低剂量应用于培养的白血病细胞时促进MDM2降解和p53激活,并在小鼠异种移植模型中引起肿瘤消退。最近又开发了基于Nutlin的MDM2降解酶,包括一个 MDM2同源PROTAC,其自我二聚化导致其自身泛素化和破坏。靶向截短的p53尽管大多数癌症相关的TP53突变是错义突变,约占10 %的TP53突变的肿瘤携带无义突变,产生截短的蛋白质,通常这些截断的蛋白在翻译后不久就被降解。由于这些蛋白质的寿命很短,而且往往缺乏大量的p53蛋白序列,试图通过上面描述的方法来重新激活它们,可能是毫无意义的。人们因此提出了两种可供选择的方法来激活存在p53截短突变的癌细胞中的p53信号通路,第一种方法是使翻译机器能够绕过RNA终止密码子并产生全长p53蛋白。这些化合物包括氨基糖苷类抗生素庆大霉素及其衍生物,如G418和新一代合成衍生物NB124。使用这些氨基糖苷类药物治疗可以帮助完整p53的合成,促进癌细胞凋亡;另一种方法一种补充的方法需要抑制无义介导的mRNA降解(NMD)过程。其中一个例子是NMD14,它的目标是NMD 机制中的重要组件SMG7的一个结构口袋,类似的药物ataluren,已经达到了囊性纤维化的III期临床试验。它们作为抗癌药物的有效性,特别是在含有TP53无义突变的肿瘤中,仍有待确定。此外,这些化合物巨大的毒性阻碍了它们作为选择性p53靶向药物的潜力。 靶向功能增强p53突变体尽管大多数基于p53的药物研发努力都是针对恢复癌细胞中野生型p53活性,但也有尝试通过靶向p53突变体快速降解来消除突变体p53的 GOF活性。这依赖于TP53突变的癌细胞对其突变体p53蛋白上瘾的假设,HSP90热休克蛋白减弱了p53突变体的降解。亚历山德罗夫等人的研究表明,长期抑制HSP90增加了携带p53突变体表达肿瘤的小鼠的存活,而不是那些没有p53的肿瘤。帕德马纳班等鉴定了一个小分子( MCB613),它选择性地驱动p53 ( R175H )热点突变体的核输出和溶酶体降解,而不是其他突变体。虽然通过靶向消除突变体p53的 GOF仍然是一个明智的方法,但随着恢复p53功能的更好药物的出现,它可能会失去吸引力。 基于p53的肿瘤免疫治疗近年来,由于在几种类型的癌症中取得了前所未有的成功,肿瘤免疫治疗方案引起了极大的兴奋。肿瘤免疫治疗的复兴也引发了人们对基于p53的策略的重新兴趣,主要是那些旨在提高免疫系统识别和消除含有失调p53的癌细胞的能力。这种策略可能有效的预期主要是基于携带TP53错义突变的癌细胞往往过度表达p53,因此可能会通过主要组织相容性复合物( MHC )在其表面显示更多的p53衍生肽。一般来说,癌细胞中错义的p53蛋白的过度表达会增加各种肽的表达,这些肽来自于所有的p53蛋白。虽然这些多肽中至少有一部分可能与p53共享,但免疫系统对癌细胞的选择性将依赖于正常细胞只表达极低量的p53 (图4 )。这种方法的可行性得到了T细胞对p53的反应不受天然自我耐受限制这一观察结果的支持。然而,健康细胞不受影响的假设是有风险的:的确,分化的细胞可能具有极低的p53 mRNA含量,因此几乎不合成任何p53蛋白,但这可能不适用于快速增殖的正常祖细胞,其中TP53 mRNA表达量更多。 基于p53的疫苗疫苗接种的尝试,旨在提高细胞免疫力,对抗含有大量过量p53的癌细胞。在这些尝试中使用的肽的序列来自于p53蛋白的区域,这些区域在癌症中很少发生突变,因此与癌症相关的突变体p53共享。然而,癌细胞的选择性是由于正常细胞具有非常低的p53含量,因此不能被接种疫苗的宿主的免疫系统识别和攻击。随后,一种合成的长肽(SLP)疫苗,包括来自p53序列的10个重叠肽,每3周注射两次,显示在转移性结直肠癌中引起以CD4 + T细胞为主的T细胞反应。不良事件相对较轻:毒性局限于1/2级,多发生在接种部位处。在卵巢癌患者在SLP疫苗接种之前,低剂量环磷酰胺治疗增强了p53的免疫原性。然而,一项临床试验未能显示SLP疫苗接种对历史对照的益处。一种编码p53的改良痘苗病毒安卡拉(MVA)疫苗也在难治性胃肠道肿瘤和卵巢癌患者的早期临床试验中进行了测试,在总共11例患者中,分别有6例和5例患者能够诱导CD8+和CD4+T细胞应答。
重要的是,p53疫苗接种后出现免疫应答的患者与CD8+T细胞无扩增的患者相比,无进展生存期显著延长。使用MVAp53疫苗和抗PD1抗体帕博利珠单抗的进一步临床试验目前正在进行中( NCT03113487,NCT02432963 )。树突状细胞被修饰以表达MHCⅠ类和Ⅱ上的p53多肽。这种DC - p53疫苗在16例SCLC患者中触发了p53特异性免疫反应,其中28例患者接受了治疗。重要的是,在接种p53疫苗后接受二次化疗的21名患者中,13名患者表现出期望的临床反应。令人失望的是,紫II期随机试验DC-p53疫苗或对照显示总体反应率没有差异。
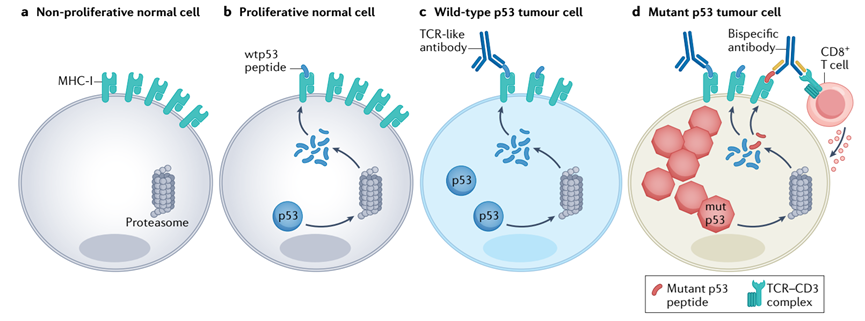
2019冠状病毒疾病(COVID-19)大流行期间mRNA疫苗接种的成功为p53 mRNA疫苗带来了新的希望。在引入自体TP53 mRNA转染后在乳腺癌患者的DC中,18例高表达p53的肿瘤患者中有13例表现出p53特异性γ干扰素T细胞反应;p53的特异性IFN γ T细胞应答在 健康人和p53低表达的乳腺癌患者间形成了鲜明对比。这种方法很可能会随着最近的先进方法而复兴。P53特异性抗体其他基于p53的免疫治疗方法也在不断出现。T细胞受体模拟物( T cell receptor mimic,TCRm )抗体,也称为TCR样抗体,是一种潜在的靶向细胞内蛋白的策略。这些抗体通常由噬菌体展示文库筛选或杂交瘤筛选产生,识别MHC I类分子在细胞表面展示的抗原表位,类似于T细胞通过TCR识别这些抗原表位,从而识别来自细胞内蛋白质的多肽(图4)。
因此,Li等开发了一种新的TCRm抗体,该抗体识别由p53衍生的抗原表位,该表位被癌细胞选择性地显示在MHC I类上,而不是正常的外周血单核细胞。重要的是,这种p53 TCRm抗体在乳腺癌移植瘤小鼠中引起了肿瘤消退。同样,Low等产生了一种p53特异性TCR样抗体,命名为P1C1TM。P1C1TM是基于p53多肽设计的,但它对含有几种不同p53突变的癌细胞具有选择性的抗体依赖的细胞毒性,这可能是由于它们的高p53丰度。作为一种额外的治疗优势,P1C1TM还可以通过抗体-药物偶联物促进药物递送到p53突变的癌细胞中。
p53和肿瘤微环境
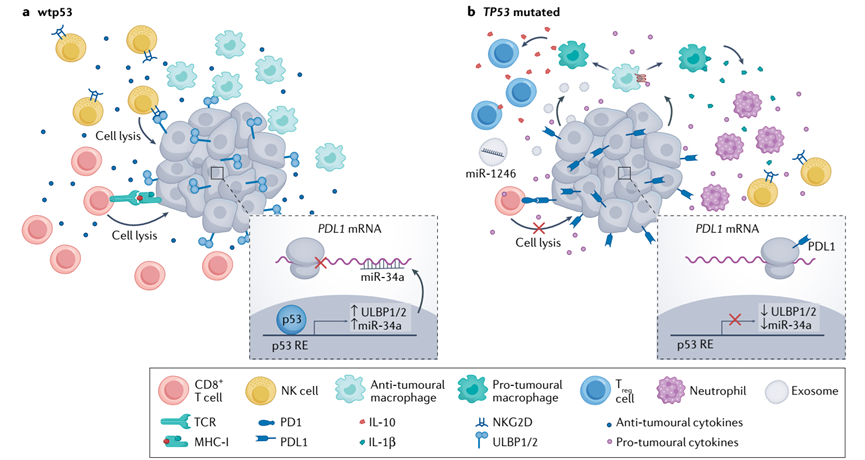
除了有针对性地尝试开发p53特异性免疫治疗方法之外,最近的研究强调了p53和肿瘤免疫治疗之间更广泛的联系。因此,p53在癌细胞中的状态可以影响TME的免疫景观(图5);在癌细胞中,有功能的p53倾向于癌症限制性的肿瘤微环境,而p53的缺失则倾向于癌症支持的肿瘤微环境。此外,一些错义的p53蛋白,作为其GOF活动的一部分,可能进一步限制免疫系统攻击癌细胞的能力。例如,p53可以通过上调miR-34a148间接降低PDL1的水平,并诱导自然杀伤(NK)细胞活化配体ULBP1和ULBP2的表达。
基于p53的基因治疗
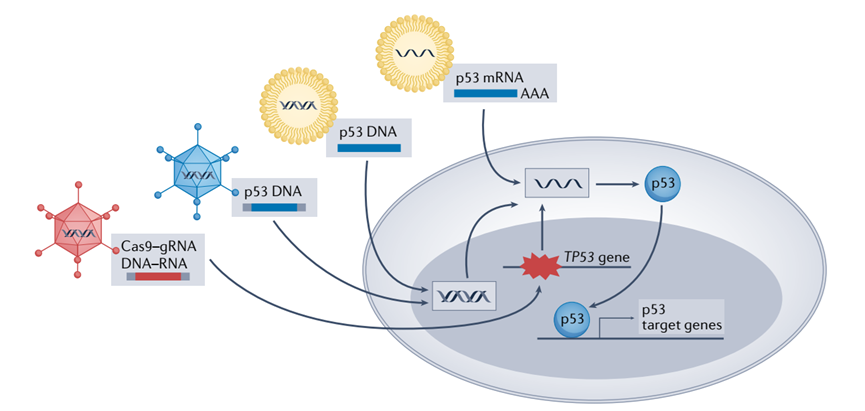
近年来,基因治疗的热情再度高涨。值得注意的是,第一个被批准用于临床的基因治疗是基于p53的。重组人p53腺病毒注射液(重组人p53腺病毒)是由深圳思必奥基因科技有限公司研发的重组人p53腺病毒,于2003年被中国食品药品监督管理总局( CFDA )批准上市用于治疗HNSCC。自那时以来,重组人p53腺病毒注射液(重组人p53腺病毒)已在中国为数以千计的患者提供治疗,据报告,在与化疗或放疗相结合的情况下,患者的反应率大大高于护理标准。
挑战和问题
尽管在以p53为基础的癌症治疗方面取得了不断的进展,但许多挑战仍然存在,寻找高效和选择性的药物最终能够进入临床仍在进行中。与所有其他抗癌治疗一样,基于p53的治疗的一个主要问题是耐药性的出现。对MDM2抑制剂耐药的一个明显机制是TP53基因的突变,这在实验上已经被证明是通过长时间的Nutlin处理。其他包括抗凋亡基因的激活,内在的抗凋亡和上调MDM4。这可能在多大程度上降低基于p53的药物的疗效仍有待观察。
此外,以p53为基础的药物不太可能作为单一疗法进入临床。许多研究试图确定有希望的相关药物组合。例如,在野生型p53 的AML模型中,将BET抑制剂与Nutlin-3a结合具有叠加效应,该模型涉及BET抑制剂增强p53的激活。BCL-2抑制剂venetoclax联合idasanutlin在原代AML细胞中取得了很好的结果,通过降低凋亡阈值它克服了对任何一种单药治疗的固有耐药。这一组合随后进入临床试验,包括正在进行的青年人和儿童复发/难治性白血病和实体瘤试验( NCT04029688 )。此外,如前所述,将p53激活与免疫治疗相结合也很有吸引力。这种联合治疗可能会减少所需的剂量,甚至在某些情况下可以克服抗药性。
另一个令人关注的问题是,基于p53的药物的体内测试主要是在老鼠身上进行的。尽管小鼠模型仍然是药物发现的标准工具,但小鼠与人类之间存在许多差异,包括p53、MDM2和MDM4蛋白序列的种间差异,以及p53信号通路的差异。
最后,也许最大的挑战是我们对人类生物学的不完全理解和对药物作用下癌细胞内发生的复杂过程的不完全理解。因此,尽管我们的筛选方法在不断改进,并产生了越来越多的p53靶向化合物,但我们仍然缺乏足够的知识来预测可靠的脱靶效应,甚至是不受欢迎的靶效应,从而导致了不受欢迎的情况,即只有在给患者服用药物时才会发现不可接受的毒性。因此,迫切需要更深入的理解和更确定的检验模型。总结过去三十年来,人们一直在努力开发基于p53的疗法,虽然成效甚微,但是靶向p53的成功绝对会彻底改变癌症治疗,这保证了靶向p53的努力不会停止。靶向p53的药物研发无疑是一项高风险同时高收益的事业。近年来,人们逐渐摒弃了p53不可药物治疗的观点,但在p53靶向治疗能够成为标准治疗方案的一部分之前,仍有许多工作有待探索。也许可以从最近KRAS抑制的令人兴奋的发展中得到鼓励, KRAS此前也因为一直失败的尝试,被认为是不可药靶点。 从众多失败中总结的经验是,没有靶向p53的单一的万能药物,正如单一的KRAS (G12C)抑制剂不会使癌症患者受益于其他KRAS突变的患者一样。因此,谨慎的患者-药物匹配将至关重要。幸运的是,由于p53功能失调在人类癌症中的普遍存在,所以即使只在一小部分p53功能失调的病例中显示出疗效的方案也能使大量患者受益。




